Subhash Santhilal, Andersson Per-Ola, Kosalai Subazini Thankaswamy, Kanduri Chandrasekhar, Kanduri Meena
Department of Medical Genetics, Institute of Biomedicine, Sahlgrenska Academy, Gothenburg University, Gothenburg, Sweden.
Department of Internal Medicine and Clinical Nutrition, Institute of Medicine Sahlgrenska Academy, Gothenburg University, Gothenburg, Sweden ; Department of Internal Medicine, Södra Älvsborg Hospital, Borås, Sweden.
Clin Epigenetics. 2016 Oct 12;8:106. doi: 10.1186/s13148-016-0274-6. eCollection 2016.
Methyl-CpG-binding domain protein enriched genome-wide sequencing (MBD-Seq) is a robust and powerful method for analyzing methylated CpG-rich regions with complete genome-wide coverage. In chronic lymphocytic leukemia (CLL), the role of CpG methylated regions associated with transcribed long noncoding RNAs (lncRNA) and repetitive genomic elements are poorly understood. Based on MBD-Seq, we characterized the global methylation profile of high CpG-rich regions in different CLL prognostic subgroups based on IGHV mutational status.
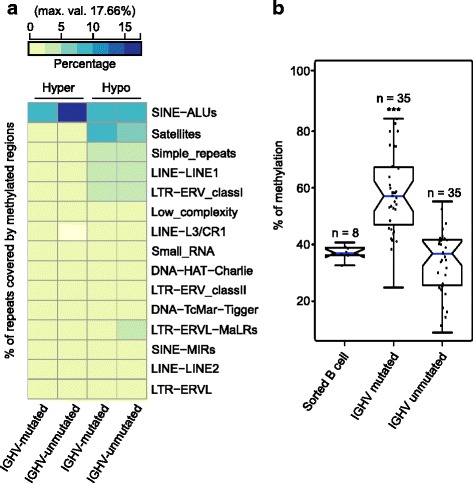
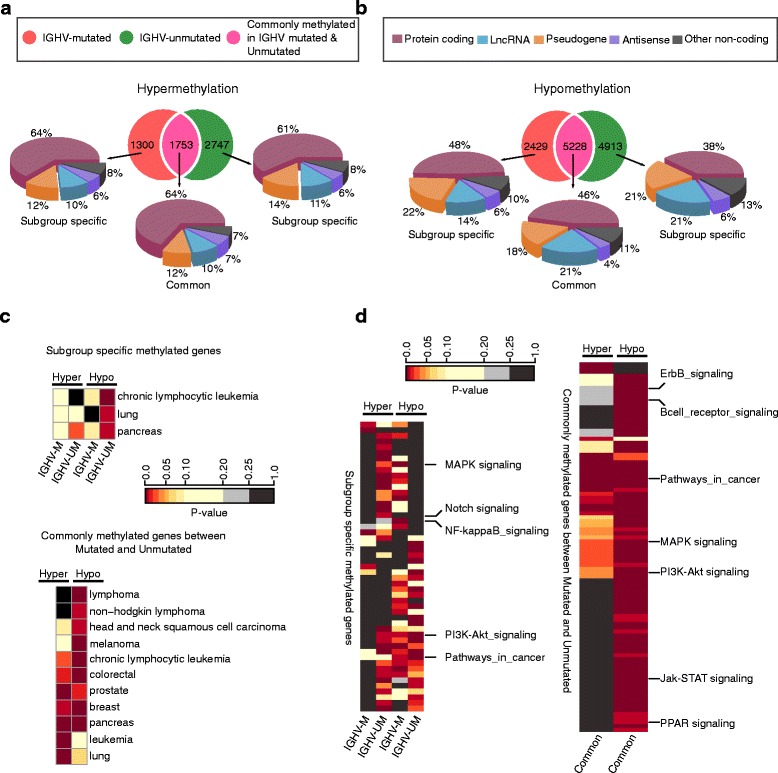
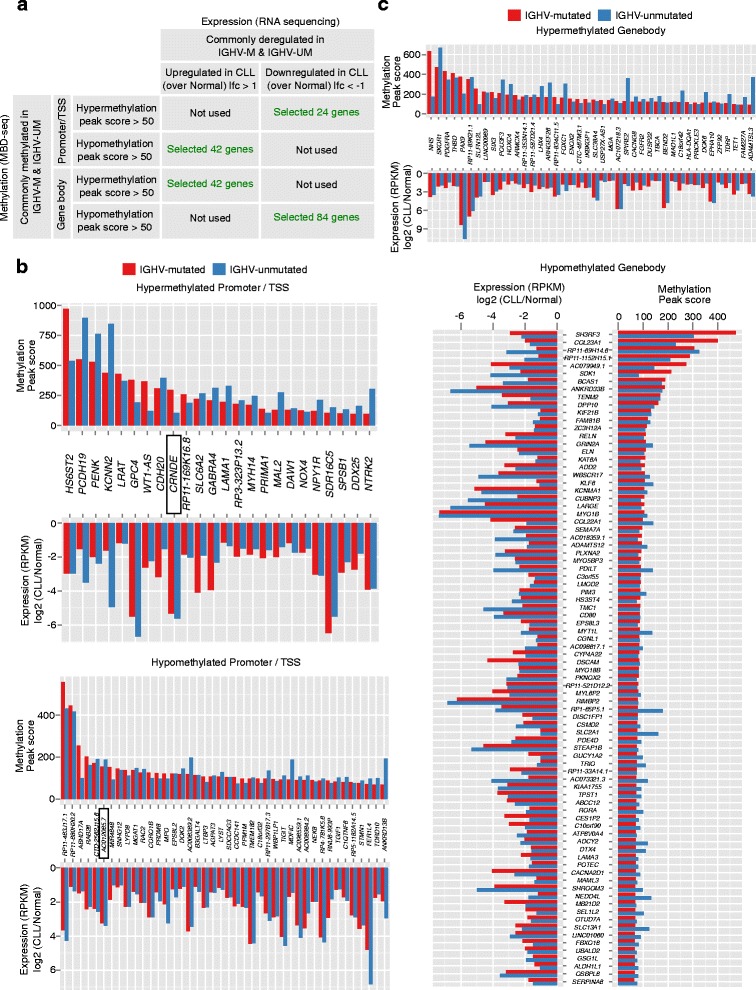
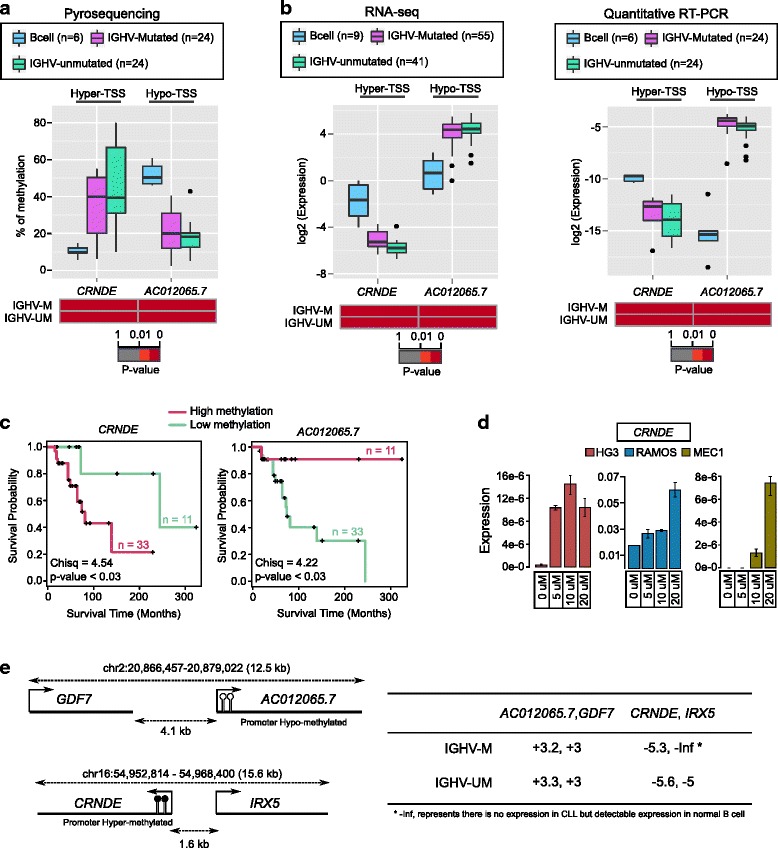
Our study identified 5800 hypermethylated and 12,570 hypomethylated CLL-specific differentially methylated genes (cllDMGs) compared to normal controls. From cllDMGs, 40 % of hypermethylated and 60 % of hypomethylated genes were mapped to noncoding RNAs. In addition, we found that the major repetitive elements such as short interspersed elements (SINE) and long interspersed elements (LINE) have a high percentage of cllDMRs (differentially methylated regions) in IGHV subgroups compared to normal controls. Finally, two novel lncRNAs (hypermethylated and hypomethylated ) were validated in an independent CLL sample cohort (48 samples) compared with 6 normal sorted B cell samples using quantitative pyrosequencing analysis. The methylation levels showed an inverse correlation to gene expression levels analyzed by real-time quantitative PCR. Notably, survival analysis revealed that hypermethylation of and hypomethylation of correlated with an inferior outcome.
Thus, our comprehensive methylation analysis by MBD-Seq provided novel hyper and hypomethylated long noncoding RNAs, repetitive elements, along with protein coding genes as potential epigenetic-based CLL-signature genes involved in disease pathogenesis and prognosis.
全基因组甲基化结合结构域蛋白富集测序(MBD-Seq)是一种强大且有效的方法,可用于分析具有全基因组完整覆盖的富含甲基化CpG的区域。在慢性淋巴细胞白血病(CLL)中,与转录的长链非编码RNA(lncRNA)和重复性基因组元件相关的CpG甲基化区域的作用尚不清楚。基于MBD-Seq,我们根据IGHV突变状态对不同CLL预后亚组中富含CpG的区域的整体甲基化谱进行了特征分析。
与正常对照相比,我们的研究鉴定出5800个高甲基化和12570个低甲基化的CLL特异性差异甲基化基因(cllDMGs)。在cllDMGs中,40%的高甲基化基因和60%的低甲基化基因被定位到非编码RNA。此外,我们发现,与正常对照相比,主要的重复元件,如短散在元件(SINE)和长散在元件(LINE)在IGHV亚组中具有高比例的cllDMRs(差异甲基化区域)。最后,使用定量焦磷酸测序分析,在一个独立的CLL样本队列(48个样本)与6个正常分选的B细胞样本中验证了两个新的lncRNAs(高甲基化的 和低甲基化的 )。甲基化水平与通过实时定量PCR分析的基因表达水平呈负相关。值得注意的是,生存分析显示, 的高甲基化和 的低甲基化与较差的预后相关。
因此,我们通过MBD-Seq进行的全面甲基化分析提供了新的高甲基化和低甲基化的长链非编码RNA、重复元件以及蛋白质编码基因,作为参与疾病发病机制和预后的潜在基于表观遗传学的CLL特征基因。