Swiderski Kristy, Thakur Savant S, Naim Timur, Trieu Jennifer, Chee Annabel, Stapleton David I, Koopman René, Lynch Gordon S
Basic and Clinical Myology Laboratory, Department of Physiology, The University of Melbourne, Melbourne, 3010 Australia.
Skelet Muscle. 2016 Oct 24;6:36. doi: 10.1186/s13395-016-0108-4. eCollection 2016.
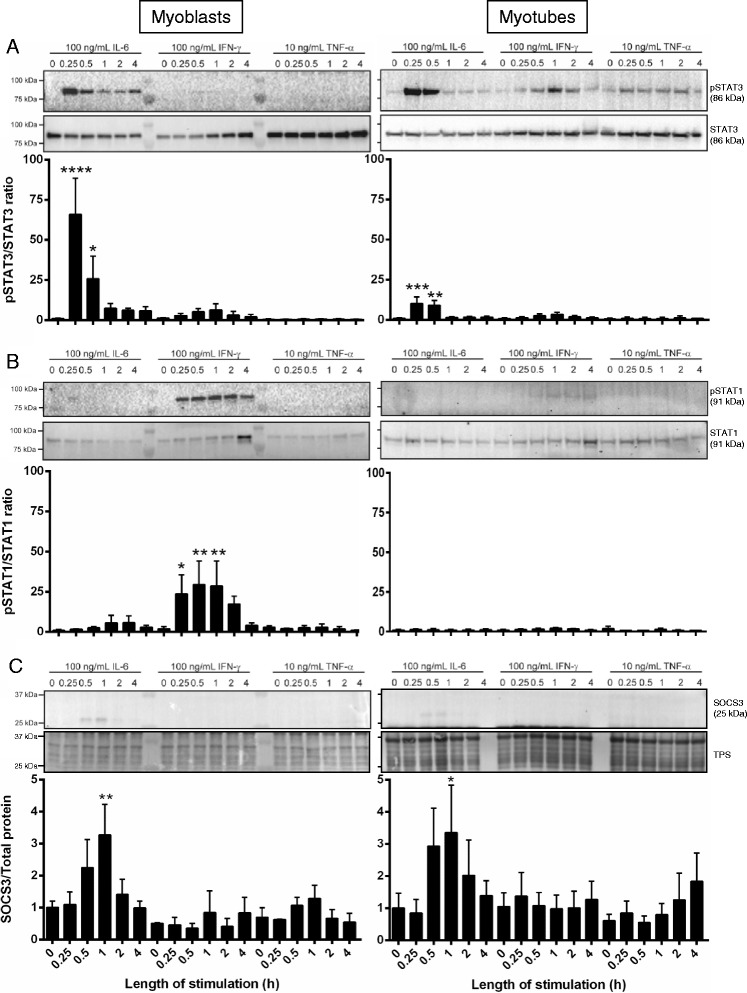
Muscles of old animals are injured more easily and regenerate poorly, attributed in part to increased levels of circulating pro-inflammatory cytokines. The Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling cascade is a key mediator of inflammatory cytokine action, and signaling via this pathway is increased in muscles with aging. As a negative regulator of JAK/STAT signaling, a key mediator of myogenic proliferation and differentiation, altered expression of suppressor of cytokine signaling (SOCS3) is likely to have important consequences for muscle regeneration. To model this scenario, we investigated the effect of SOCS3 deletion within mature muscle fibers on injury and repair. We tested the hypothesis that reduced SOCS3 function would alter the inflammatory response and impair muscle regeneration after myotoxic injury.
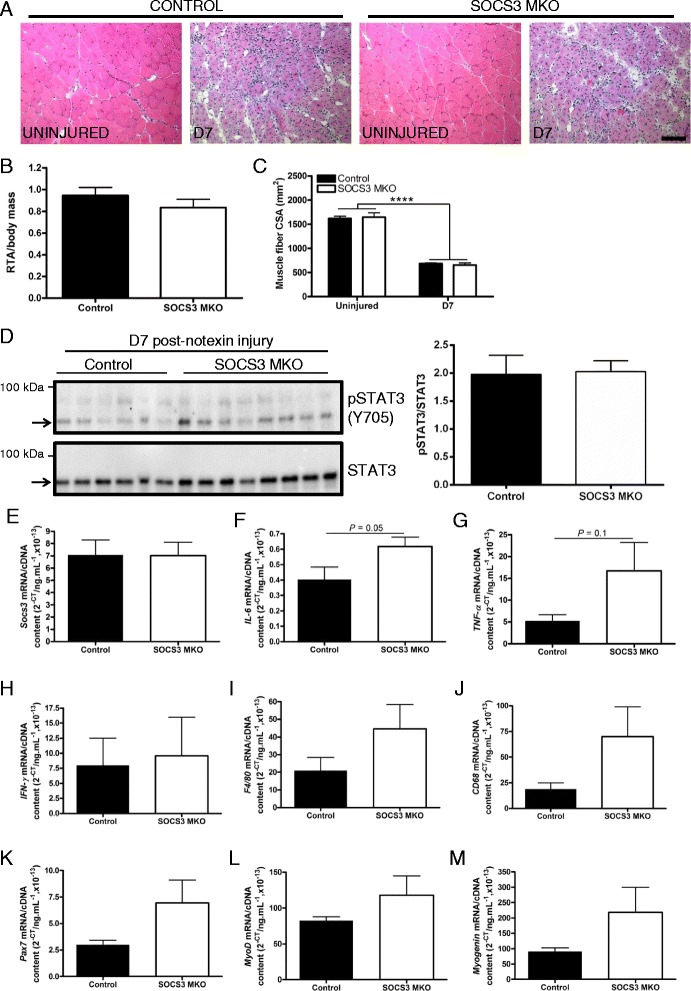
Mice with a specific deletion of SOCS3 within mature skeletal muscle fibers were used to assess the effect of SOCS3 deletion on muscle injury and repair. Twelve-week-old or 24-month-old SOCS3 muscle-specific knockout (SOCS3 MKO) mice and littermate controls were either left uninjured or injured with a single injection of notexin (10 μg/ml) into the right tibialis anterior (TA) muscle. At 1, 2, 3, 5, 7, or 14 days post-injury, the right TA muscle was excised and subjected to histological, western immunoblotting, and gene expression analyses. Force production and fatigue were assessed in uninjured muscles and at 7 days post-notexin injury.
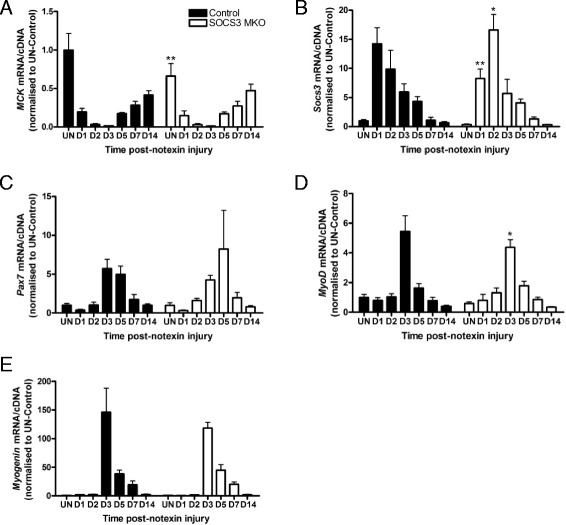
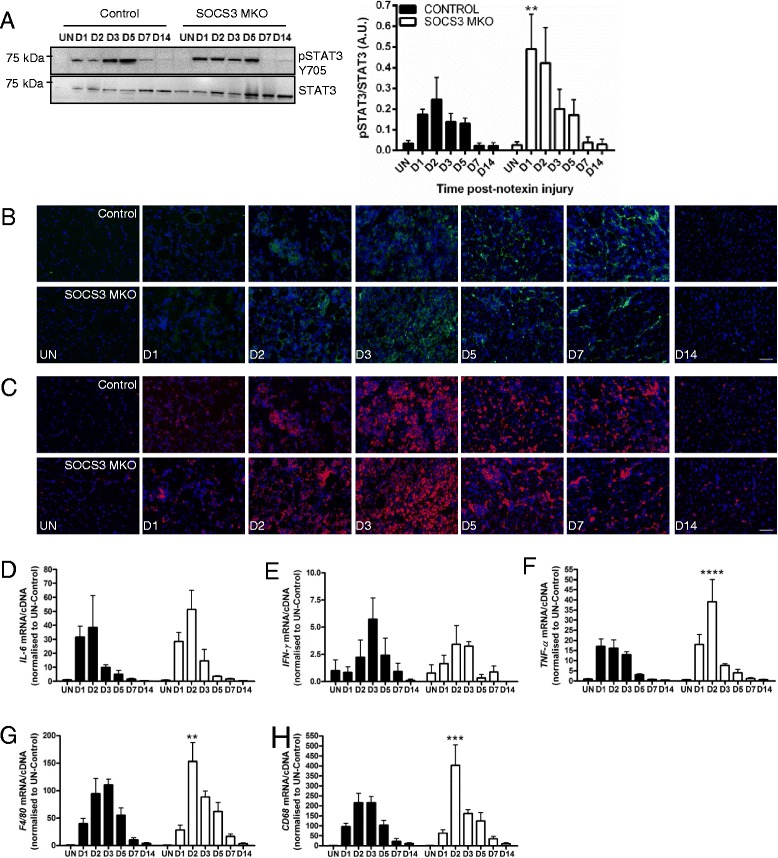
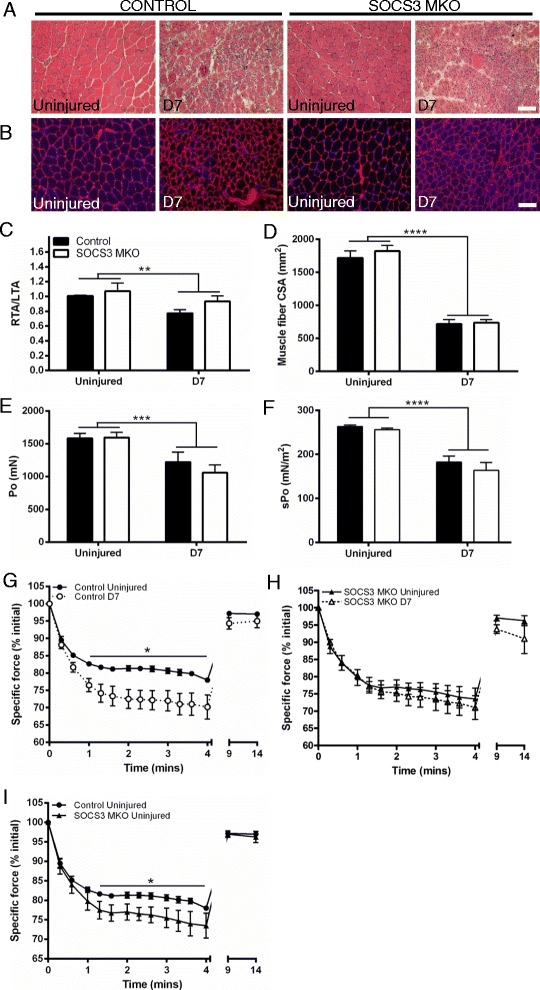
In uninjured muscles, SOCS3 deletion decreased force production during fatigue but had no effect on the gross or histological appearance of the TA muscles. After notexin injury, deletion of SOCS3 increased STAT3 phosphorylation at day 1 and increased the mRNA expression of the inflammatory cytokine , and the inflammatory cell markers and at day 2. Gene expression analysis of the regeneration markers , , and indicated SOCS3 deletion had no effect on the progression of muscle repair after notexin injury. Inflammation and regeneration were also unchanged in the muscles of 24-month-old SOCS3 MKO mice compared with control.
Loss of SOCS3 expression in mature muscle fibers increased the inflammatory response to myotoxic injury but did not impair muscle regeneration in either adult or old mice. Therefore, reduced SOCS3 expression in muscle fibers is unlikely to underlie impaired muscle regeneration. Further investigation into the role of SOCS3 in other cell types involved in muscle repair is warranted.
老年动物的肌肉更容易受伤且再生能力差,部分原因是循环中促炎细胞因子水平升高。Janus激酶/信号转导子和转录激活子(JAK/STAT)信号级联是炎性细胞因子作用的关键介质,并且随着肌肉衰老,通过该途径的信号传导会增加。作为JAK/STAT信号传导的负调节因子,细胞因子信号转导抑制因子3(SOCS3)是肌源性增殖和分化的关键介质,其表达改变可能对肌肉再生产生重要影响。为模拟这种情况,我们研究了成熟肌纤维中SOCS3缺失对损伤和修复的影响。我们检验了以下假设:SOCS3功能降低会改变炎性反应并损害肌毒素损伤后的肌肉再生。
使用在成熟骨骼肌纤维中特异性缺失SOCS3的小鼠来评估SOCS3缺失对肌肉损伤和修复的影响。12周龄或24月龄的SOCS3肌肉特异性敲除(SOCS3 MKO)小鼠及其同窝对照,要么不进行损伤处理,要么通过向右侧胫前肌(TA)单次注射诺维毒素(10μg/ml)进行损伤。在损伤后1、2、3、5、7或14天,切除右侧TA肌肉并进行组织学、蛋白质免疫印迹和基因表达分析。在未损伤的肌肉以及诺维毒素损伤后7天评估肌力产生和疲劳情况。
在未损伤的肌肉中,SOCS3缺失会降低疲劳期间的肌力产生,但对TA肌肉的大体外观或组织学表现没有影响。诺维毒素损伤后,SOCS3缺失在第1天增加了STAT3磷酸化,并在第2天增加了炎性细胞因子以及炎性细胞标志物和的mRNA表达。对再生标志物、和的基因表达分析表明,SOCS3缺失对诺维毒素损伤后肌肉修复的进程没有影响。与对照相比,24月龄SOCS3 MKO小鼠肌肉中的炎症和再生也没有变化。
成熟肌纤维中SOCS3表达缺失增加了对肌毒素损伤的炎性反应,但在成年或老年小鼠中均未损害肌肉再生。因此,肌纤维中SOCS3表达降低不太可能是肌肉再生受损的原因。有必要进一步研究SOCS3在参与肌肉修复的其他细胞类型中的作用。