Kinjo Akira R, Nakamura Haruki
Institute for Protein Research, Osaka University, Suita, Osaka, 565-0871, Japan.
Biophysics (Nagoya-shi). 2007 Dec 28;3:75-84. doi: 10.2142/biophysics.3.75. eCollection 2007.
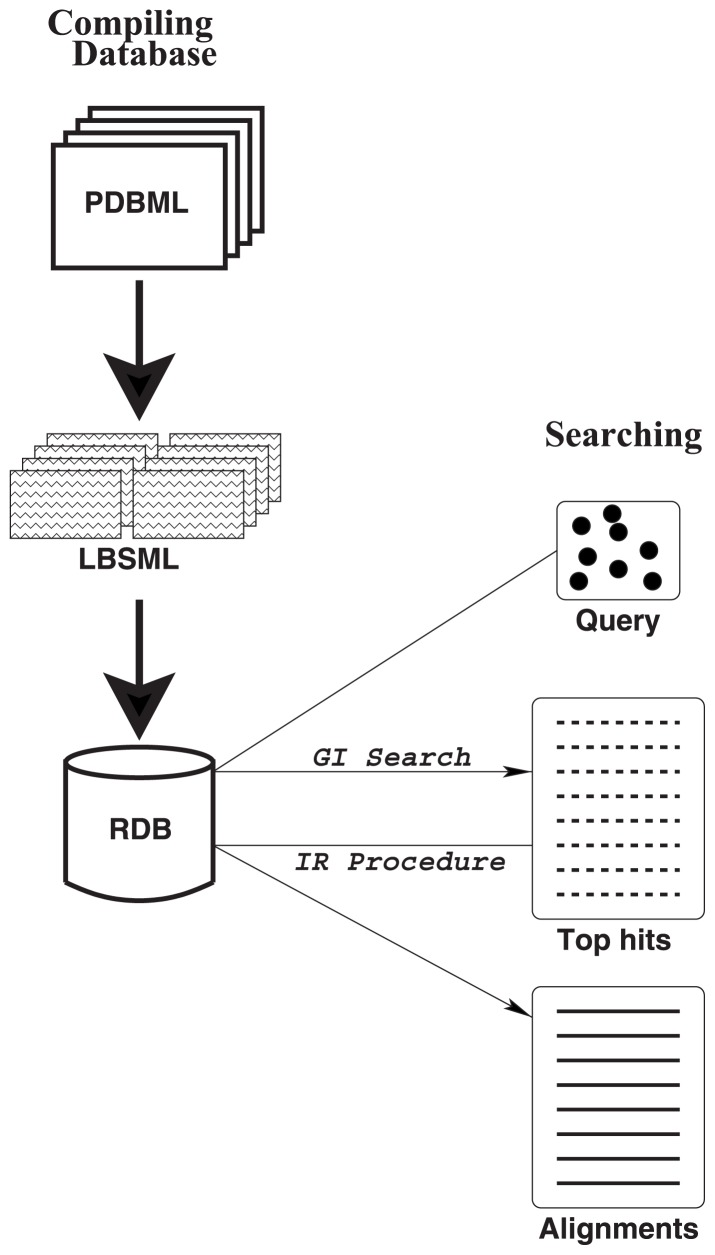
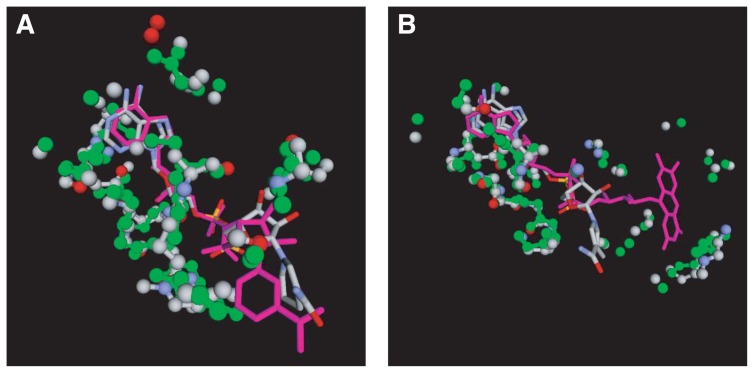
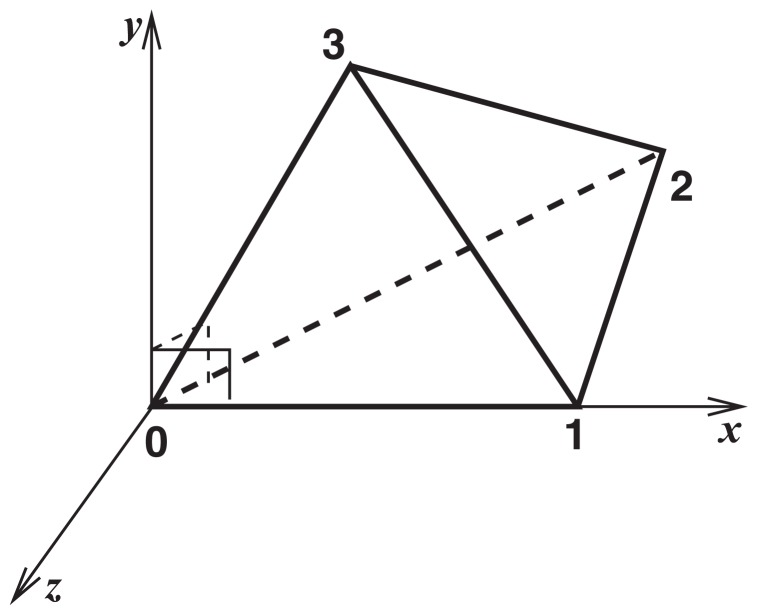
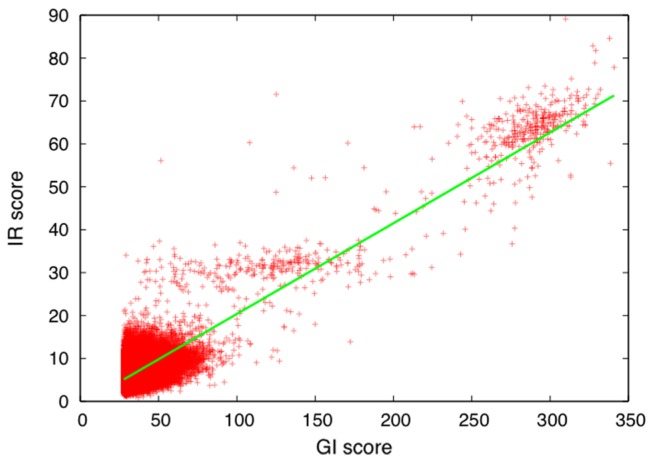
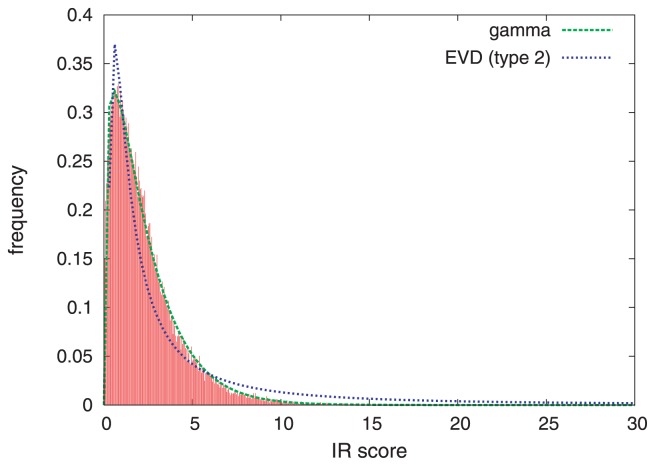
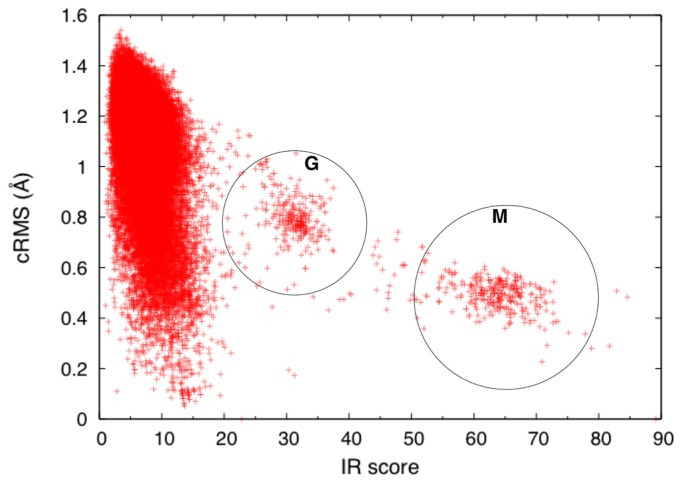
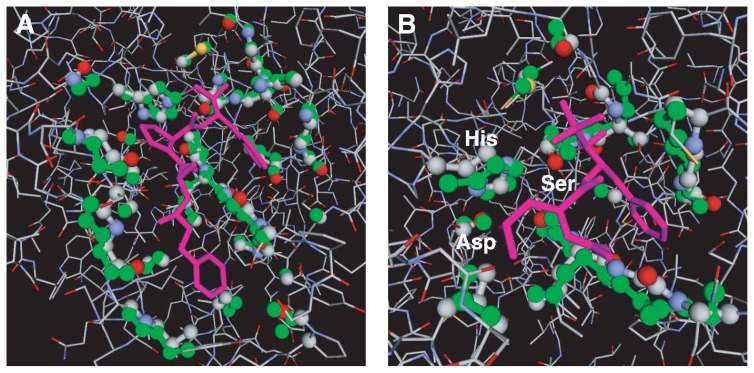
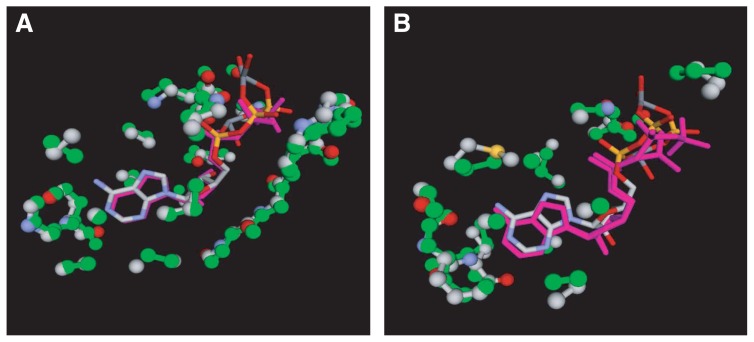
A method to search for local structural similarities in proteins at atomic resolution is presented. It is demonstrated that a huge amount of structural data can be handled within a reasonable CPU time by using a conventional relational database management system with appropriate indexing of geometric data. This method, which we call geometric indexing, can enumerate ligand binding sites that are structurally similar to sub-structures of a query protein among more than 160,000 possible candidates within a few hours of CPU time on an ordinary desktop computer. After detecting a set of high scoring ligand binding sites by the geometric indexing search, structural alignments at atomic resolution are constructed by iteratively applying the Hungarian algorithm, and the statistical significance of the final score is estimated from an empirical model based on a gamma distribution. Applications of this method to several protein structures clearly shows that significant similarities can be detected between local structures of non-homologous as well as homologous proteins.
本文提出了一种在原子分辨率下搜索蛋白质局部结构相似性的方法。结果表明,通过使用具有适当几何数据索引的传统关系数据库管理系统,可以在合理的CPU时间内处理大量的结构数据。我们将这种方法称为几何索引,它可以在普通台式计算机上几个小时的CPU时间内,在超过160,000个可能的候选物中枚举与查询蛋白质的子结构在结构上相似的配体结合位点。通过几何索引搜索检测到一组高分配体结合位点后,通过迭代应用匈牙利算法构建原子分辨率下的结构比对,并根据基于伽马分布的经验模型估计最终得分的统计显著性。该方法在几种蛋白质结构上的应用清楚地表明,在非同源以及同源蛋白质的局部结构之间可以检测到显著的相似性。