Gomez-Cabrero David, Almgren Malin, Sjöholm Louise K, Hensvold Aase H, Ringh Mikael V, Tryggvadottir Rakel, Kere Juha, Scheynius Annika, Acevedo Nathalie, Reinius Lovisa, Taub Margaret A, Montano Carolina, Aryee Martin J, Feinberg Jason I, Feinberg Andrew P, Tegnér Jesper, Klareskog Lars, Catrina Anca I, Ekström Tomas J
Center for Molecular Medicine at Karolinska Institutet and Karolinska University Hospital, Stockholm, Sweden.
Department of Medicine, Unit of Computational Medicine, Stockholm, Sweden.
Genome Med. 2016 Nov 22;8(1):124. doi: 10.1186/s13073-016-0374-0.
Twin studies are powerful models to elucidate epigenetic modifications resulting from gene-environment interactions. Yet, commonly a limited number of clinical twin samples are available, leading to an underpowered situation afflicted with false positives and hampered by low sensitivity. We investigated genome-wide DNA methylation data from two small sets of monozygotic twins representing different phases during the progression of rheumatoid arthritis (RA) to find novel genes for further research.
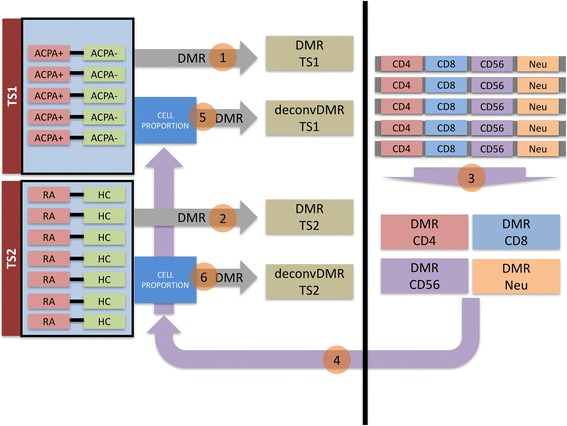
We implemented a robust statistical methodology aimed at investigating a small number of samples to identify differential methylation utilizing the comprehensive CHARM platform with whole blood cell DNA from two sets of twin pairs discordant either for ACPA (antibodies to citrullinated protein antigens)-positive RA versus ACPA-negative healthy or for ACPA-positive healthy (a pre-RA stage) versus ACPA-negative healthy. To deconvolute cell type-dependent differential methylation, we assayed the methylation patterns of sorted cells and used computational algorithms to resolve the relative contributions of different cell types and used them as covariates.
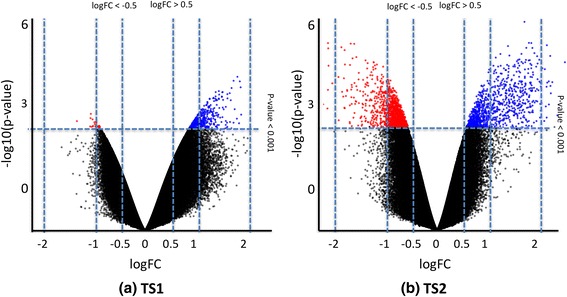
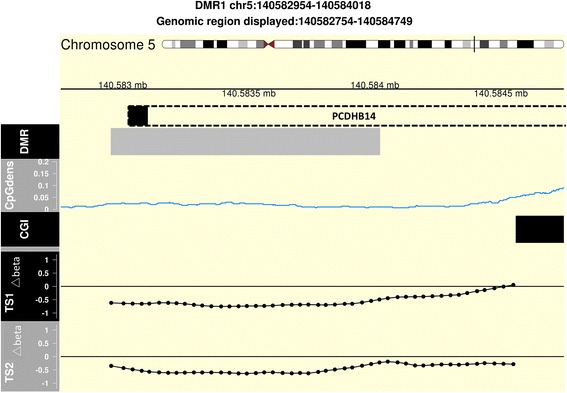
To identify methylation biomarkers, five healthy twin pairs discordant for ACPAs were profiled, revealing a single differentially methylated region (DMR). Seven twin pairs discordant for ACPA-positive RA revealed six significant DMRs. After deconvolution of cell type proportions, profiling of the healthy ACPA discordant twin-set revealed 17 genome-wide significant DMRs. When methylation profiles of ACPA-positive RA twin pairs were adjusted for cell type, the analysis disclosed one significant DMR, associated with the EXOSC1 gene. Additionally, the results from our methodology suggest a temporal connection of the protocadherine beta-14 gene to ACPA-positivity with clinical RA.
Our biostatistical methodology, optimized for a low-sample twin design, revealed non-genetically linked genes associated with two distinct phases of RA. Functional evidence is still lacking but the results reinforce further study of epigenetic modifications influencing the progression of RA. Our study design and methodology may prove generally useful in twin studies.
双胞胎研究是阐明基因 - 环境相互作用所导致的表观遗传修饰的有力模型。然而,通常可获得的临床双胞胎样本数量有限,导致出现功效不足的情况,存在假阳性问题且灵敏度较低。我们研究了来自两组小样本单卵双胞胎的全基因组DNA甲基化数据,这两组双胞胎代表类风湿性关节炎(RA)进展过程中的不同阶段,以寻找可供进一步研究的新基因。
我们实施了一种稳健的统计方法,旨在利用综合CHARM平台,对少量样本进行研究,以识别差异甲基化情况。该平台使用了两组双胞胎对的全血细胞DNA,这两组双胞胎对在抗环瓜氨酸肽(ACPA,即抗瓜氨酸化蛋白抗原的抗体)阳性的RA与ACPA阴性的健康状态之间存在差异,或者在ACPA阳性的健康状态(RA前期)与ACPA阴性的健康状态之间存在差异。为了反卷积细胞类型依赖性差异甲基化,我们检测了分选细胞的甲基化模式,并使用计算算法来解析不同细胞类型的相对贡献,并将其用作协变量。
为了识别甲基化生物标志物,对五对ACPA状态不同的健康双胞胎进行了分析,发现了一个差异甲基化区域(DMR)。七对ACPA阳性RA状态不同的双胞胎发现了六个显著的DMR。在对细胞类型比例进行反卷积后,对ACPA状态不同的健康双胞胎组的分析揭示了17个全基因组显著的DMR。当对ACPA阳性RA双胞胎对的甲基化谱进行细胞类型调整后,分析发现了一个与EXOSC1基因相关的显著DMR。此外,我们方法的结果表明原钙黏蛋白β - 14基因与ACPA阳性以及临床RA之间存在时间上的联系。
我们针对低样本双胞胎设计优化的生物统计学方法,揭示了与RA两个不同阶段相关的非遗传连锁基因。虽然仍缺乏功能证据,但结果加强了对影响RA进展的表观遗传修饰的进一步研究。我们的研究设计和方法可能在双胞胎研究中普遍有用。