Mieth Bettina, Kloft Marius, Rodríguez Juan Antonio, Sonnenburg Sören, Vobruba Robin, Morcillo-Suárez Carlos, Farré Xavier, Marigorta Urko M, Fehr Ernst, Dickhaus Thorsten, Blanchard Gilles, Schunk Daniel, Navarro Arcadi, Müller Klaus-Robert
Machine Learning Group, Technische Universität Berlin, Berlin, 10587, Germany.
Department of Computer Science, Humboldt University of Berlin, Berlin, 10099, Germany.
Sci Rep. 2016 Nov 28;6:36671. doi: 10.1038/srep36671.
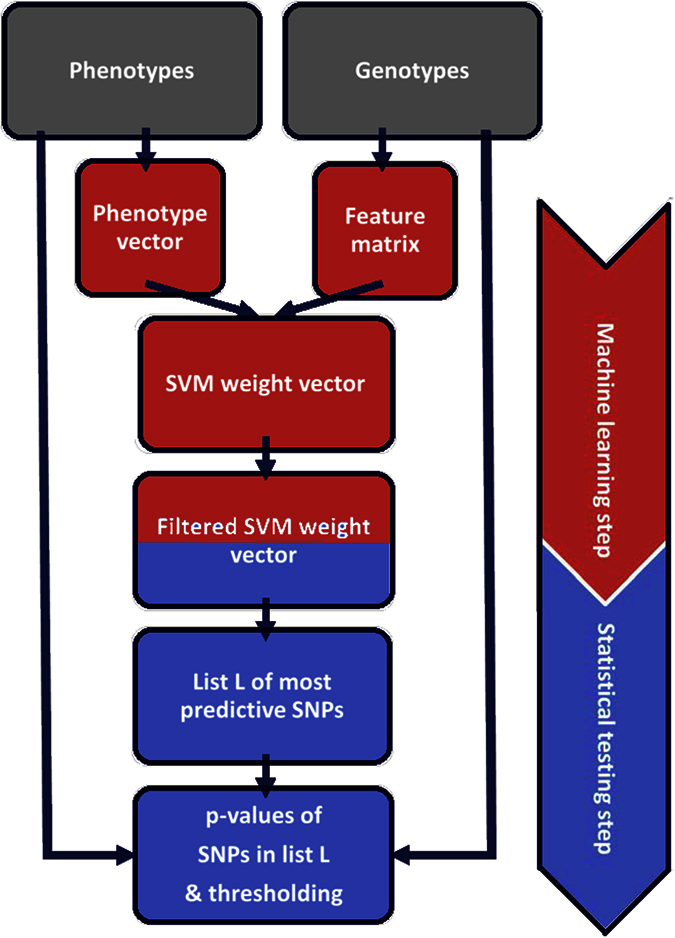
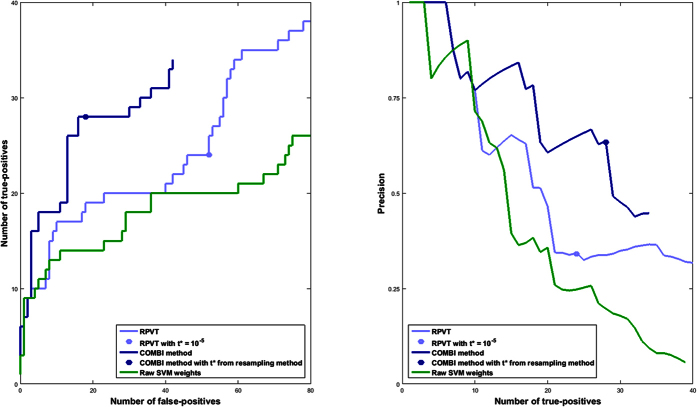
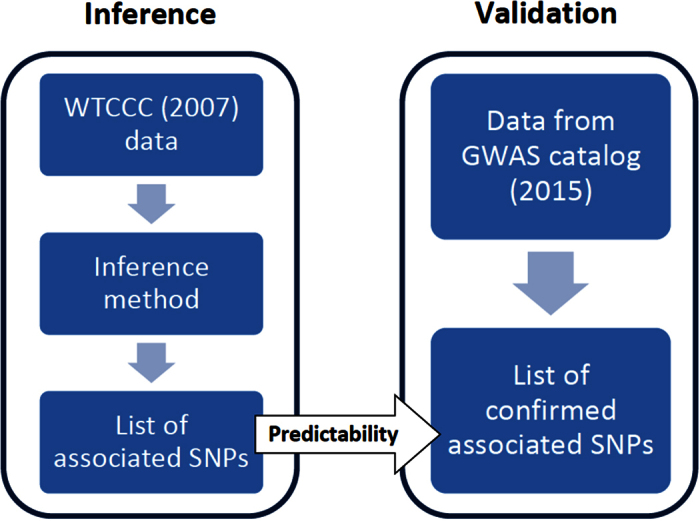
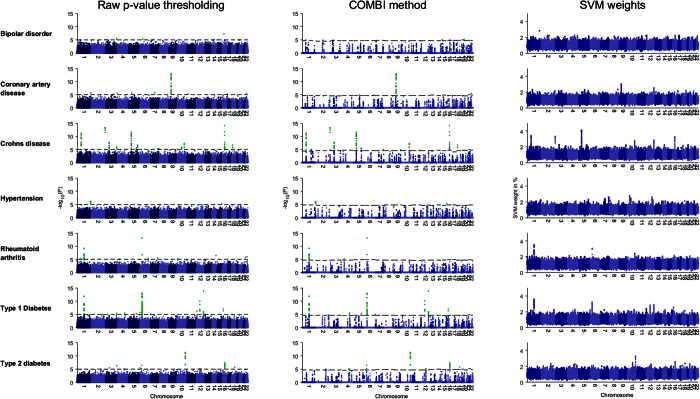
The standard approach to the analysis of genome-wide association studies (GWAS) is based on testing each position in the genome individually for statistical significance of its association with the phenotype under investigation. To improve the analysis of GWAS, we propose a combination of machine learning and statistical testing that takes correlation structures within the set of SNPs under investigation in a mathematically well-controlled manner into account. The novel two-step algorithm, COMBI, first trains a support vector machine to determine a subset of candidate SNPs and then performs hypothesis tests for these SNPs together with an adequate threshold correction. Applying COMBI to data from a WTCCC study (2007) and measuring performance as replication by independent GWAS published within the 2008-2015 period, we show that our method outperforms ordinary raw p-value thresholding as well as other state-of-the-art methods. COMBI presents higher power and precision than the examined alternatives while yielding fewer false (i.e. non-replicated) and more true (i.e. replicated) discoveries when its results are validated on later GWAS studies. More than 80% of the discoveries made by COMBI upon WTCCC data have been validated by independent studies. Implementations of the COMBI method are available as a part of the GWASpi toolbox 2.0.
全基因组关联研究(GWAS)的标准分析方法是对基因组中的每个位置单独进行测试,以确定其与所研究表型之间关联的统计学显著性。为了改进GWAS分析,我们提出了一种机器学习与统计检验相结合的方法,该方法以数学上严格可控的方式考虑了所研究的单核苷酸多态性(SNP)集合中的相关结构。这种新颖的两步算法COMBI,首先训练一个支持向量机来确定候选SNP的子集,然后对这些SNP进行假设检验,并进行适当的阈值校正。将COMBI应用于WTCCC研究(2007年)的数据,并以2008 - 2015年期间发表的独立GWAS的复制情况来衡量性能,我们发现我们的方法优于普通的原始p值阈值法以及其他现有技术方法。当在后续的GWAS研究中验证其结果时,COMBI比所检验的其他方法具有更高的功效和精度,同时产生更少的错误(即未复制的)发现和更多真实(即已复制的)发现。COMBI对WTCCC数据所做的发现中,超过80%已被独立研究验证。COMBI方法的实现可作为GWASpi工具箱2.0的一部分获取。