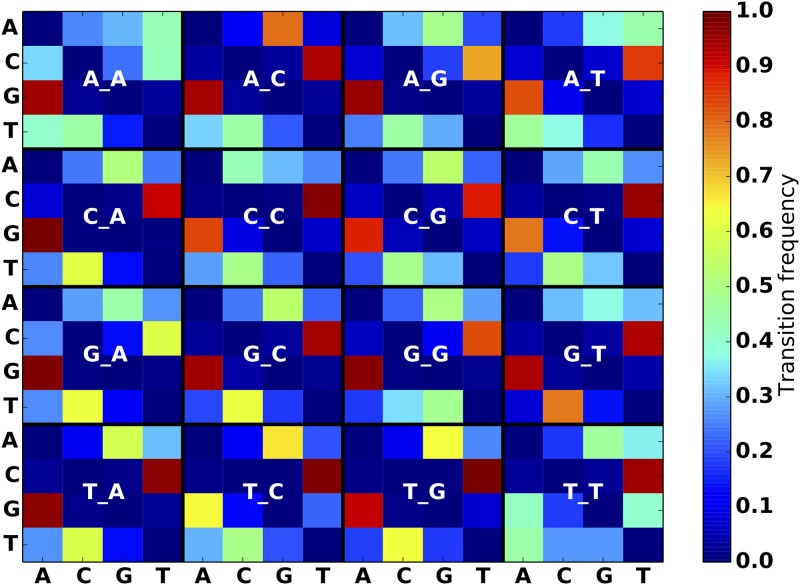
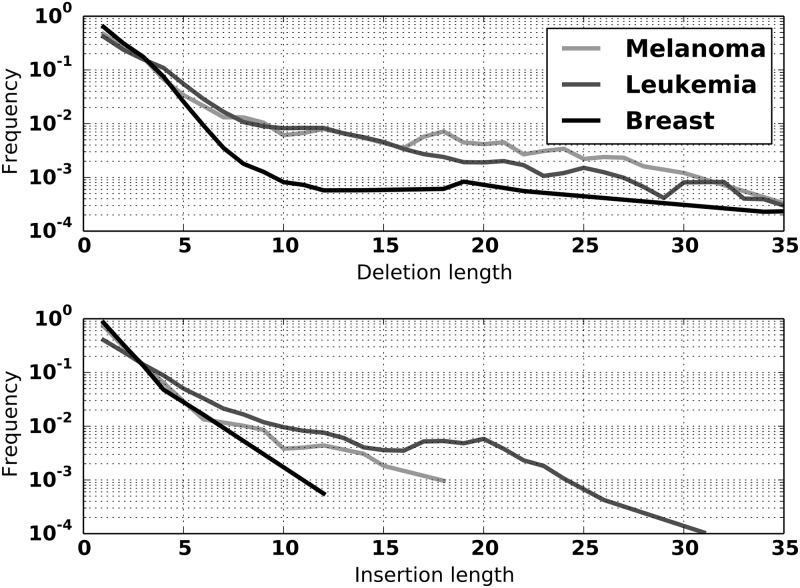
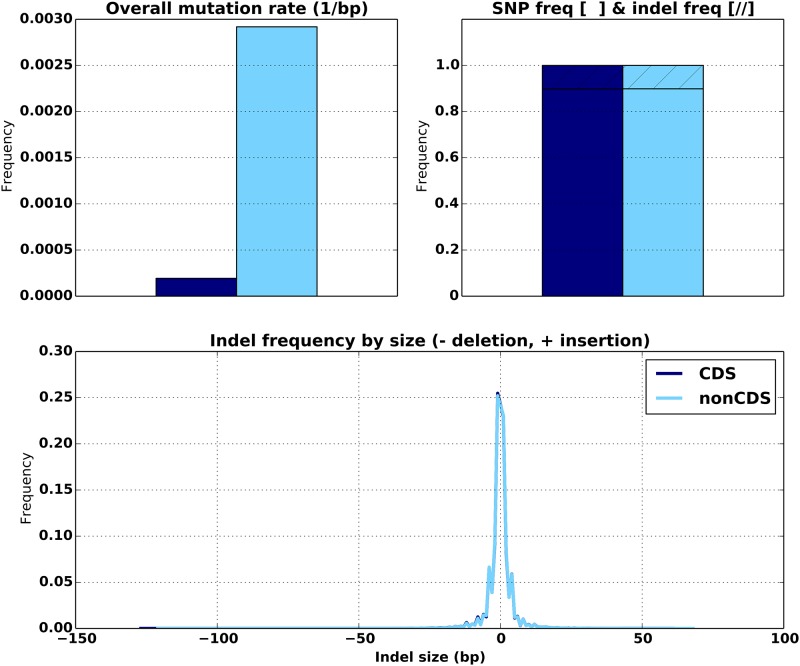
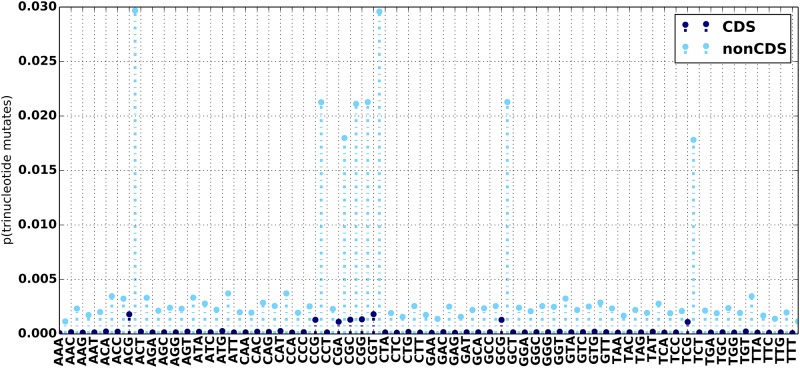
Stephens Zachary D, Hudson Matthew E, Mainzer Liudmila S, Taschuk Morgan, Weber Matthew R, Iyer Ravishankar K
Department of Electrical and Computer Engineering, Univ. of Illinois at Urbana-Champaign, Urbana, IL, United States of America.
Department of Crop Sciences, Univ. of Illinois at Urbana-Champaign, Urbana, IL, United States of America.
PLoS One. 2016 Nov 28;11(11):e0167047. doi: 10.1371/journal.pone.0167047. eCollection 2016.
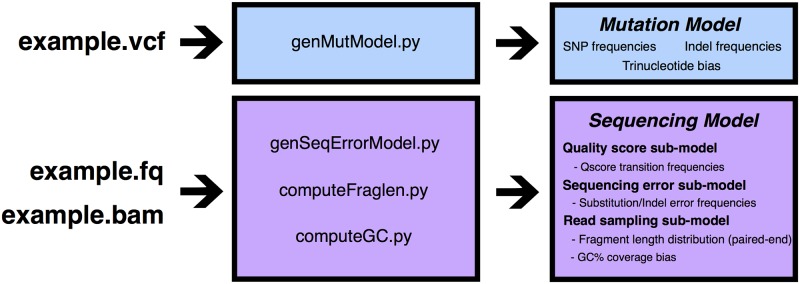
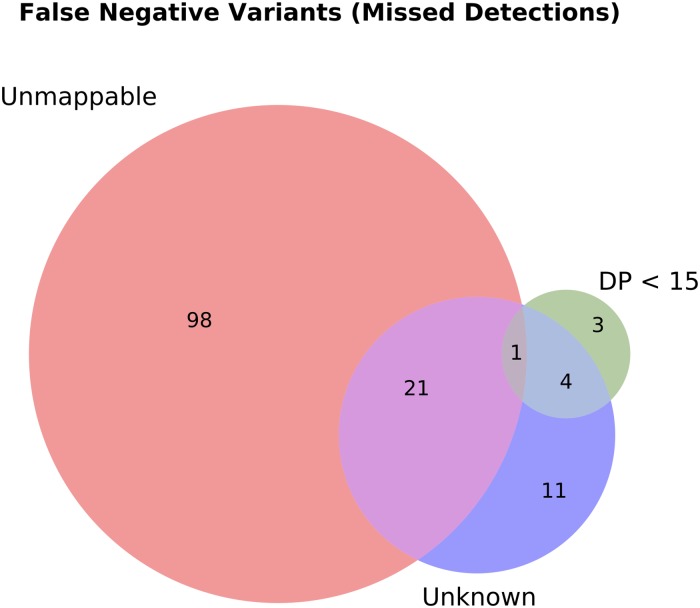
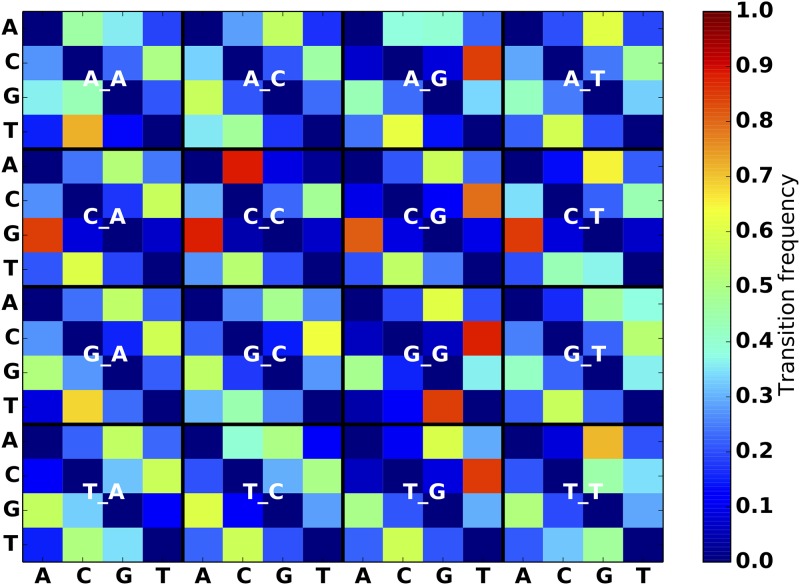
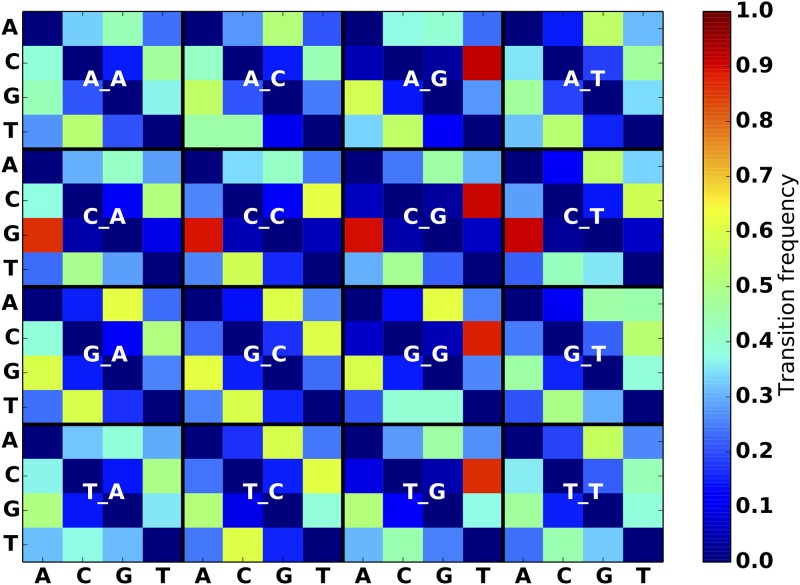
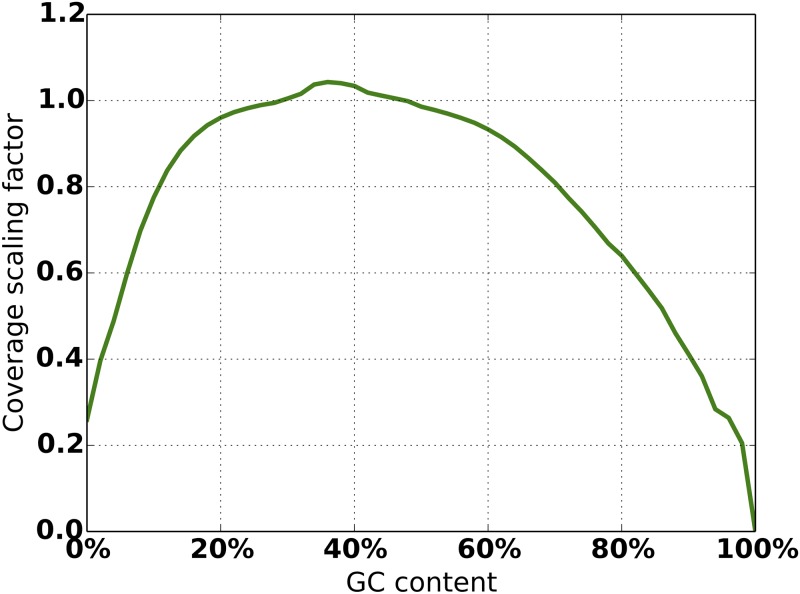
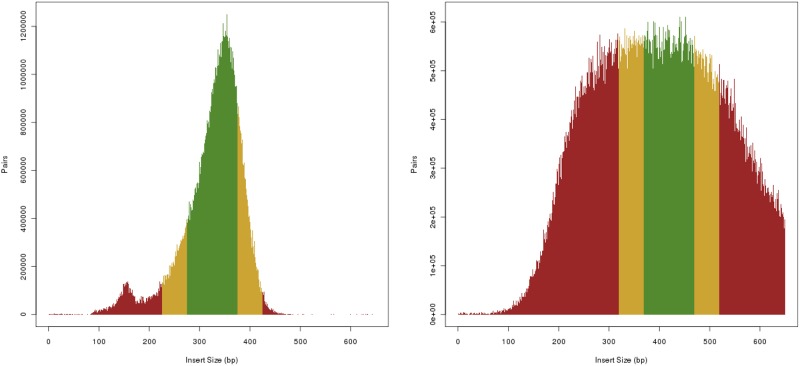
An obstacle to validating and benchmarking methods for genome analysis is that there are few reference datasets available for which the "ground truth" about the mutational landscape of the sample genome is known and fully validated. Additionally, the free and public availability of real human genome datasets is incompatible with the preservation of donor privacy. In order to better analyze and understand genomic data, we need test datasets that model all variants, reflecting known biology as well as sequencing artifacts. Read simulators can fulfill this requirement, but are often criticized for limited resemblance to true data and overall inflexibility. We present NEAT (NExt-generation sequencing Analysis Toolkit), a set of tools that not only includes an easy-to-use read simulator, but also scripts to facilitate variant comparison and tool evaluation. NEAT has a wide variety of tunable parameters which can be set manually on the default model or parameterized using real datasets. The software is freely available at github.com/zstephens/neat-genreads.
验证和基准测试基因组分析方法的一个障碍是,几乎没有可用的参考数据集,对于这些数据集,样本基因组突变图谱的“真实情况”是已知的且经过充分验证。此外,真实人类基因组数据集的免费公开可用性与保护捐赠者隐私不兼容。为了更好地分析和理解基因组数据,我们需要能够模拟所有变异的测试数据集,这些变异既能反映已知生物学特征,又能体现测序假象。读取模拟器可以满足这一要求,但常因与真实数据相似度有限以及整体灵活性不足而受到批评。我们展示了NEAT(下一代测序分析工具包),这是一组工具,不仅包括一个易于使用的读取模拟器,还包括便于变异比较和工具评估的脚本。NEAT有各种各样的可调参数,可以在默认模型上手动设置,也可以使用真实数据集进行参数化。该软件可在github.com/zstephens/neat-genreads上免费获取。