Shaheen Ranad, Szymanska Katarzyna, Basu Basudha, Patel Nisha, Ewida Nour, Faqeih Eissa, Al Hashem Amal, Derar Nada, Alsharif Hadeel, Aldahmesh Mohammed A, Alazami Anas M, Hashem Mais, Ibrahim Niema, Abdulwahab Firdous M, Sonbul Rawda, Alkuraya Hisham, Alnemer Maha, Al Tala Saeed, Al-Husain Muneera, Morsy Heba, Seidahmed Mohammed Zain, Meriki Neama, Al-Owain Mohammed, AlShahwan Saad, Tabarki Brahim, Salih Mustafa A, Faquih Tariq, El-Kalioby Mohamed, Ueffing Marius, Boldt Karsten, Logan Clare V, Parry David A, Al Tassan Nada, Monies Dorota, Megarbane Andre, Abouelhoda Mohamed, Halees Anason, Johnson Colin A, Alkuraya Fowzan S
Department of Genetics, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia.
Leeds Institute of Biomedical & Clinical Sciences, University of Leeds, Leeds, LS9 7TF, UK.
Genome Biol. 2016 Nov 28;17(1):242. doi: 10.1186/s13059-016-1099-5.
Ciliopathies are clinically diverse disorders of the primary cilium. Remarkable progress has been made in understanding the molecular basis of these genetically heterogeneous conditions; however, our knowledge of their morbid genome, pleiotropy, and variable expressivity remains incomplete.
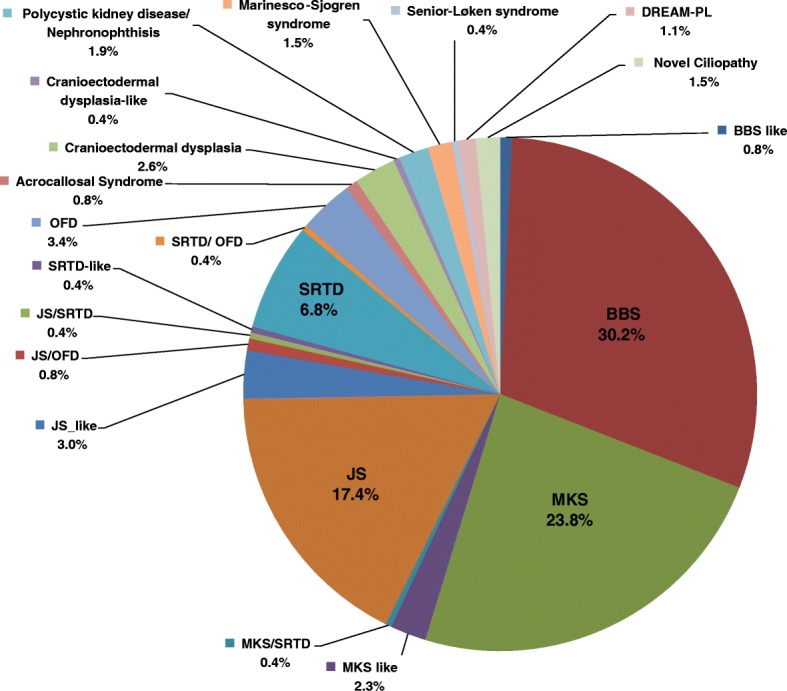
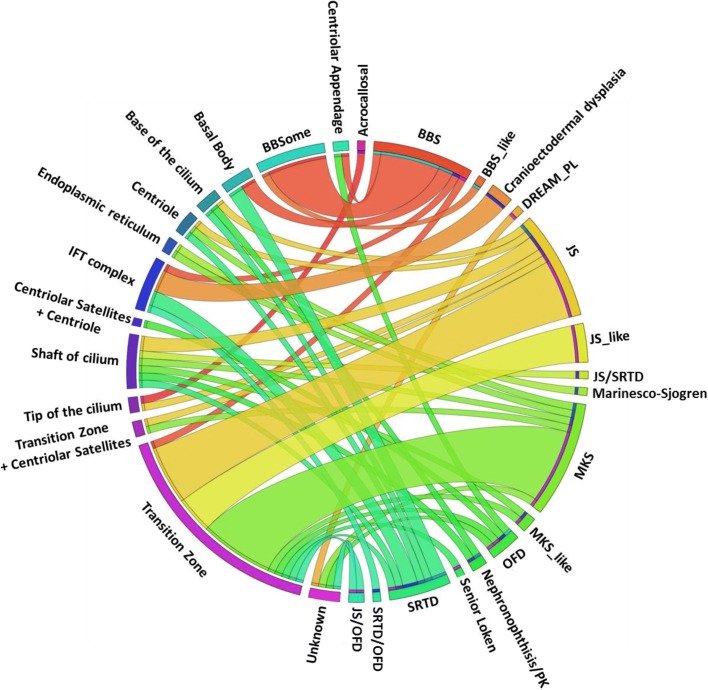
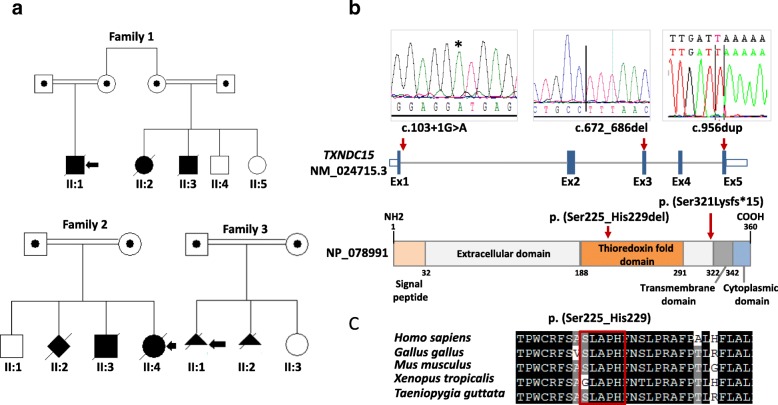
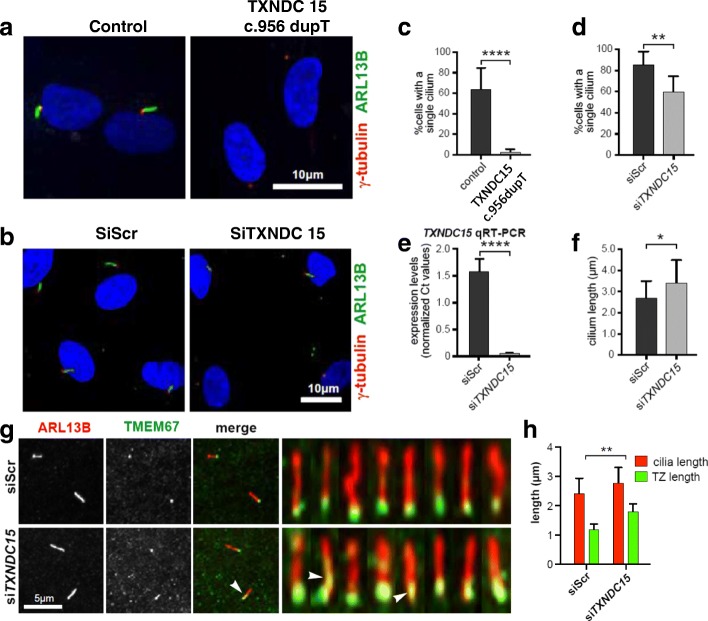
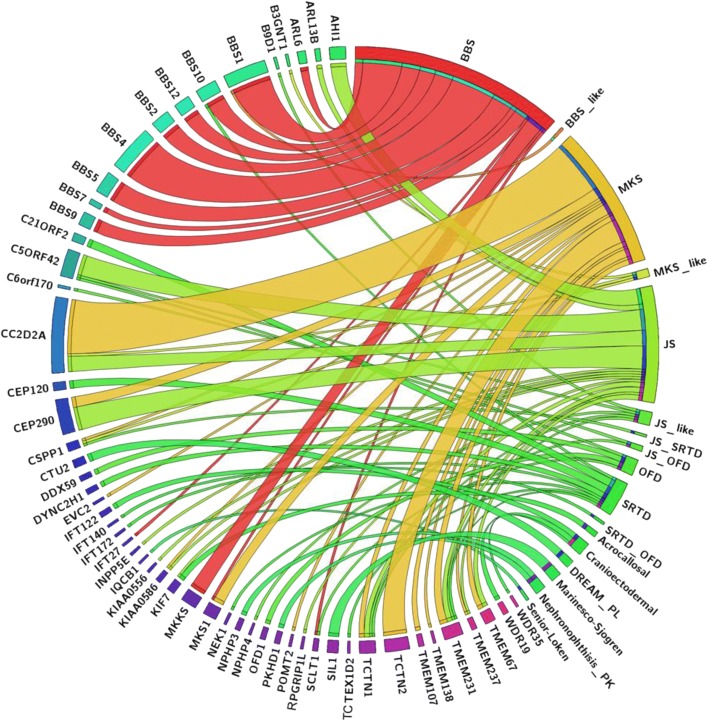
We applied genomic approaches on a large patient cohort of 371 affected individuals from 265 families, with phenotypes that span the entire ciliopathy spectrum. Likely causal mutations in previously described ciliopathy genes were identified in 85% (225/265) of the families, adding 32 novel alleles. Consistent with a fully penetrant model for these genes, we found no significant difference in their "mutation load" beyond the causal variants between our ciliopathy cohort and a control non-ciliopathy cohort. Genomic analysis of our cohort further identified mutations in a novel morbid gene TXNDC15, encoding a thiol isomerase, based on independent loss of function mutations in individuals with a consistent ciliopathy phenotype (Meckel-Gruber syndrome) and a functional effect of its deficiency on ciliary signaling. Our study also highlighted seven novel candidate genes (TRAPPC3, EXOC3L2, FAM98C, C17orf61, LRRCC1, NEK4, and CELSR2) some of which have established links to ciliogenesis. Finally, we show that the morbid genome of ciliopathies encompasses many founder mutations, the combined carrier frequency of which accounts for a high disease burden in the study population.
Our study increases our understanding of the morbid genome of ciliopathies. We also provide the strongest evidence, to date, in support of the classical Mendelian inheritance of Bardet-Biedl syndrome and other ciliopathies.
纤毛病是原发性纤毛的临床多样化疾病。在理解这些基因异质性疾病的分子基础方面已经取得了显著进展;然而,我们对其致病基因组、多效性和可变表达性的了解仍然不完整。
我们对来自265个家庭的371名受影响个体的大型患者队列应用了基因组方法,其表型涵盖了整个纤毛病谱。在85%(225/265)的家庭中鉴定出先前描述的纤毛病基因中可能的致病突变,增加了32个新的等位基因。与这些基因的完全外显模型一致,我们发现我们的纤毛病队列与对照非纤毛病队列之间,除了致病变异之外,它们的“突变负荷”没有显著差异。我们队列的基因组分析进一步确定了一个新的致病基因TXNDC15中的突变,该基因编码一种硫醇异构酶,基于具有一致纤毛病表型(梅克尔-格鲁伯综合征)个体中的独立功能丧失突变以及其缺陷对纤毛信号传导的功能影响。我们的研究还突出了七个新的候选基因(TRAPPC3、EXOC3L2、FAM98C、C17orf61、LRRCC1、NEK4和CELSR2),其中一些与纤毛发生已建立联系。最后,我们表明纤毛病的致病基因组包含许多奠基者突变,其合并携带者频率在研究人群中占很高的疾病负担。
我们的研究增进了我们对纤毛病致病基因组的理解。我们还提供了迄今为止最有力的证据,支持巴德-比埃尔综合征和其他纤毛病的经典孟德尔遗传。