Besse A, Petersen A K, Hunter J V, Appadurai V, Lalani S R, Bonnen P E
Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA.
Department of Radiology, Baylor College of Medicine and Texas Children's Hospital, Houston, TX, USA.
Mol Brain. 2016 Dec 1;9(1):93. doi: 10.1186/s13041-016-0273-8.
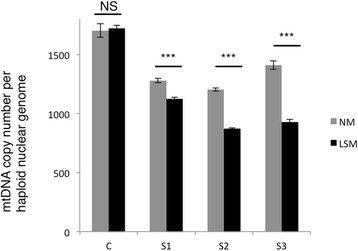
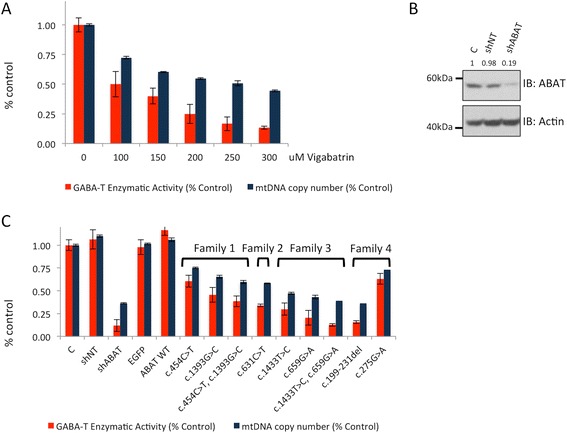
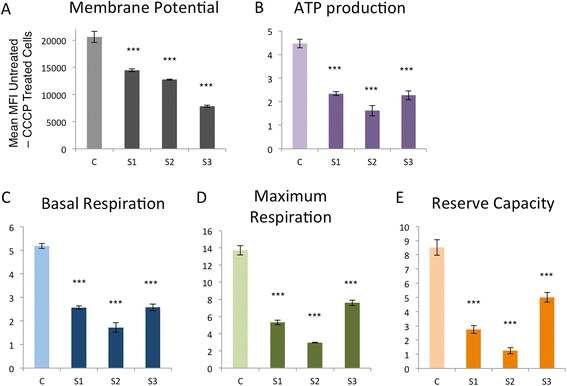
ABAT deficiency (OMIM 613163) is a rare inborn error of metabolism caused by recessive variants in the gene 4-aminobutyric acid transaminase (ABAT), which is responsible for both the catalysis of GABA and maintenance of nucleoside pools in the mitochondria. To date, only a few patients have been reported worldwide. Their clinical presentation has been remarkably consistent with primary features of severe psychomotor retardation, encephalopathy, hypotonia, and infantile-onset refractory epilepsy. We report a new case of ABAT deficiency that marks an important departure from previous clinical findings. The patient presented at age 6 months with global developmental delay, hypotonia, hypersomnolence and mild choreiform movements. At age 18 months, the subject's clinical presentation was still milder than all previously reported patients and, most notably, did not include seizures. Clinical whole exome sequencing revealed two heterozygous ABAT missense variants that are rare and predicted damaging, but never before reported in a patient and were reported as variants of unknown significance. To test the potential pathogenicity of the variants identified in this patient we developed a cell-based system to test both functions of the ABAT protein via GABA transaminase enzyme activity and mtDNA copy number assays. This systematic approach was validated using vigabatrin, the irreversible inhibitor of ABAT, and leveraged to test the functionality of all ABAT variants in previously reported patients plus the variants in this new case. This work confirmed the novel variants compromised ABAT function to similar levels as variants in previously characterized cases with more severe clinical presentation, thereby confirming the molecular diagnosis of this patient. Additionally, functional studies conducted in cells from both mild and severe patient fibroblasts showed similar levels of compromise in mitochondrial membrane potential, respiratory capacity, ATP production and mtDNA depletion. These results illustrate how cell-based functional studies can aid in the diagnosis of a rare, neurological disorder. Importantly, this patient marks an expansion in the clinical phenotype for ABAT deficiency to a milder presentation that is more commonly seen in pediatric genetics and neurology clinics.
4-氨基丁酸转氨酶(ABAT)缺乏症(OMIM 613163)是一种罕见的先天性代谢缺陷病,由4-氨基丁酸转氨酶(ABAT)基因的隐性变异引起,该基因负责催化γ-氨基丁酸(GABA)以及维持线粒体中的核苷池。迄今为止,全球仅报道了少数患者。他们的临床表现与严重精神运动发育迟缓、脑病、肌张力减退和婴儿期起病的难治性癫痫的主要特征显著一致。我们报告了一例新的ABAT缺乏症病例,该病例与先前的临床发现有重要差异。该患者6个月大时出现全面发育迟缓、肌张力减退、嗜睡和轻度舞蹈样动作。18个月大时,该患者的临床表现仍比之前报道的所有患者都要轻,最显著的是未出现癫痫发作。临床全外显子组测序发现两个杂合的ABAT错义变异,这两个变异罕见且预测具有损害性,但此前从未在患者中报道过,被报告为意义未明的变异。为了测试该患者中鉴定出的变异的潜在致病性,我们开发了一种基于细胞的系统,通过GABA转氨酶活性和线粒体DNA拷贝数测定来测试ABAT蛋白的两种功能。使用ABAT的不可逆抑制剂氨己烯酸对这种系统方法进行了验证,并利用该方法测试了先前报道患者中所有ABAT变异以及该新病例中的变异的功能。这项工作证实,这些新变异损害ABAT功能的程度与先前特征化的、临床表现更严重病例中的变异相似,从而证实了该患者的分子诊断。此外,在轻度和重度患者成纤维细胞中进行的功能研究表明,线粒体膜电位、呼吸能力、ATP产生和线粒体DNA耗竭的受损程度相似。这些结果说明了基于细胞的功能研究如何有助于诊断一种罕见的神经系统疾病。重要的是,该患者标志着ABAT缺乏症的临床表型扩展到了一种在儿科遗传学和神经科诊所更常见的较轻表现。