Gong Zizhen, Zhou Jiefei, Zhao Shengnan, Tian Chunyan, Wang Panliang, Xu Congfeng, Chen Yingwei, Cai Wei, Wu Jin
Department of Pediatric Surgery, Xinhua Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China.
Shanghai Institute for Pediatric Research, Shanghai Jiaotong University School of Medicine, Shanghai, China.
Oncotarget. 2016 Dec 20;7(51):83951-83963. doi: 10.18632/oncotarget.13796.
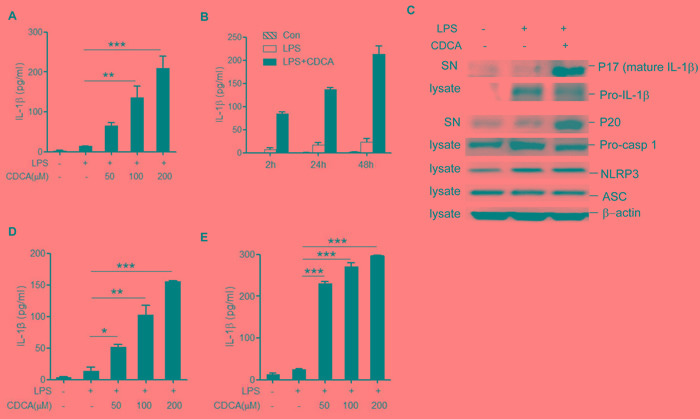
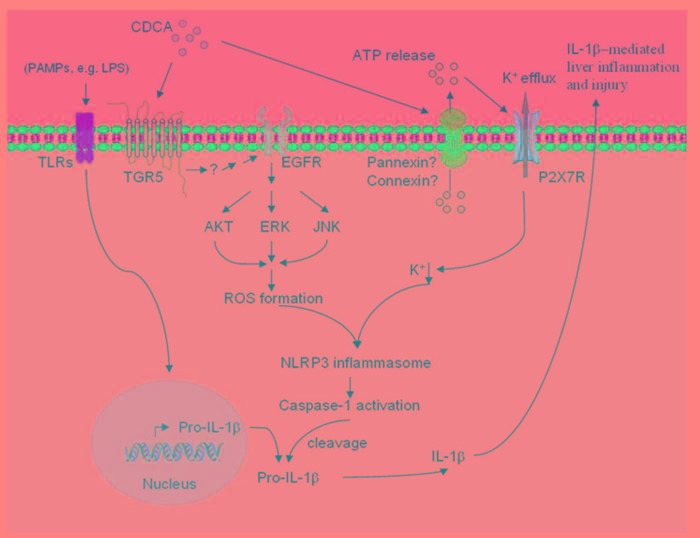
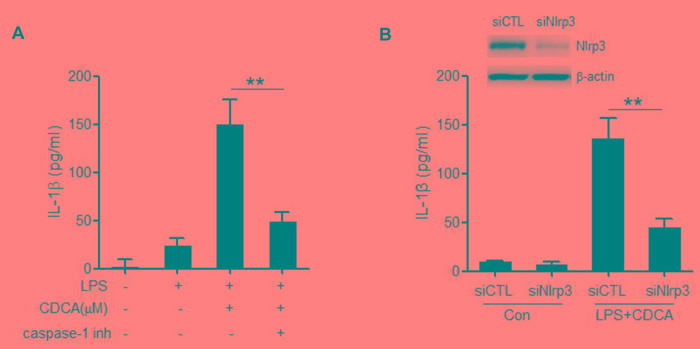
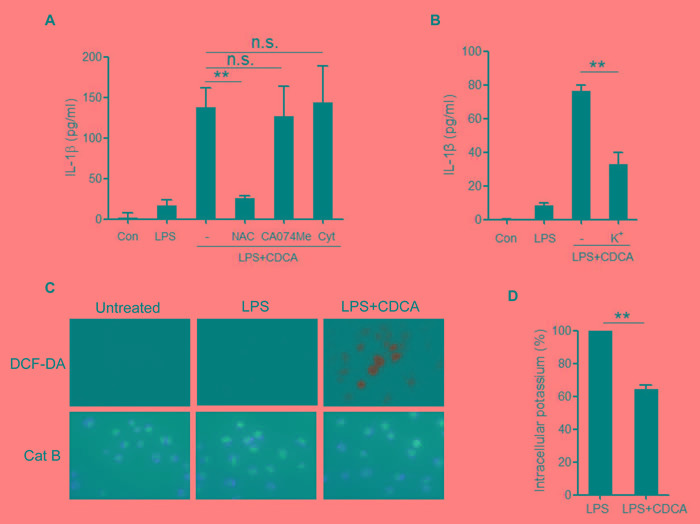
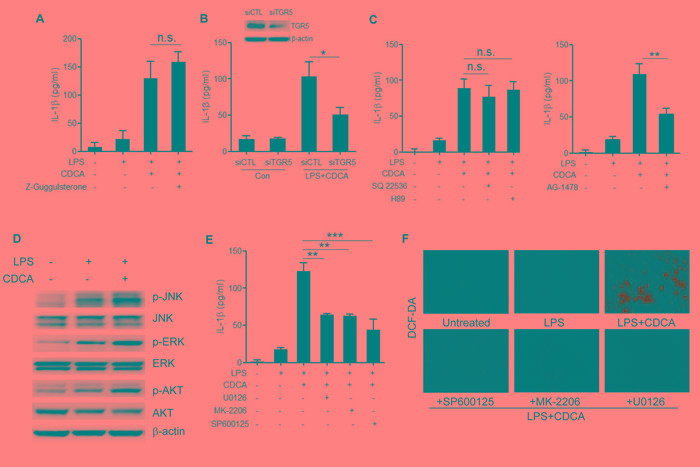
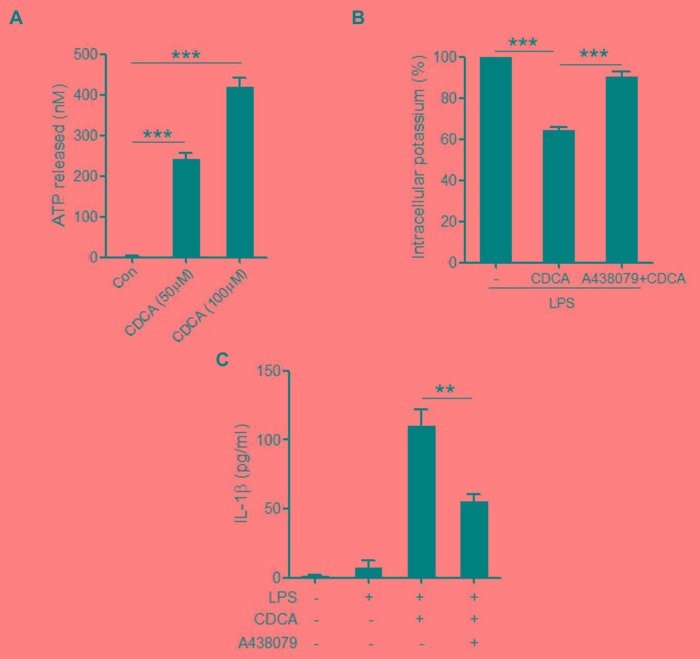
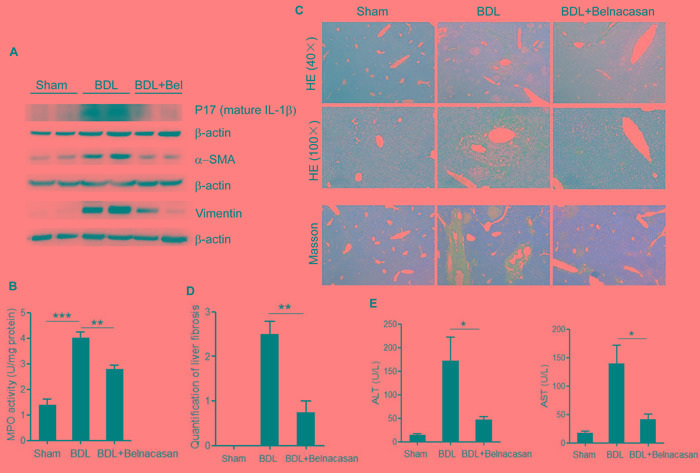
Accumulation of hydrophobic bile acids in the liver contributes to cholestatic liver injury. Inflammation induced by excessive bile acids is believed to play a crucial role, however, the mechanisms of bile acids triggered inflammatory response remain unclear. Recent studies have highlighted the effect of NLRP3 inflammasome in mediating liver inflammation and fibrosis. In this study, we for the first time showed that chenodeoxycholic acid (CDCA), the major hydrophobic primary bile acid involved in cholestatic liver injury, could dose-dependently induce NLRP3 inflammasome activation and secretion of pro-inflammatory cytokine-IL-1β in macrophages by promoting ROS production and K+ efflux. Mechanistically, CDCA triggered ROS formation in part through TGR5/EGFR downstream signaling, including protein kinase B, extracellular regulated protein kinases and c-Jun N-terminal kinase pathways. Meanwhile, CDCA also induced ATP release from macrophages which subsequently causes K+ efflux via P2X7 receptor. Furthermore, in vivo inhibition of NLRP3 inflammasome with caspase-1 inhibitor dramatically decreased mature IL-1β level of liver tissue and ameliorated liver fibrosis in bile duct ligation (BDL) mouse model. In conclusion, excessive CDCA may represent an endogenous danger signal to activate NLRP3 inflammasome and initiate liver inflammation during cholestasis. Our finding offers a mechanistic basis to ameliorate cholestatic liver fibrosis by targeting inflammasome activation.
肝脏中疏水性胆汁酸的蓄积会导致胆汁淤积性肝损伤。过量胆汁酸引发的炎症被认为起着关键作用,然而,胆汁酸触发炎症反应的机制仍不清楚。最近的研究突出了NLRP3炎性小体在介导肝脏炎症和纤维化中的作用。在本研究中,我们首次表明,鹅去氧胆酸(CDCA),即参与胆汁淤积性肝损伤的主要疏水性初级胆汁酸,可通过促进活性氧(ROS)生成和钾离子外流,在巨噬细胞中剂量依赖性地诱导NLRP3炎性小体激活和促炎细胞因子白细胞介素-1β(IL-1β)的分泌。机制上,CDCA部分通过TGR5/表皮生长因子受体(EGFR)下游信号通路触发ROS形成,包括蛋白激酶B、细胞外调节蛋白激酶和c-Jun氨基末端激酶信号通路。同时,CDCA还诱导巨噬细胞释放三磷酸腺苷(ATP),随后通过P2X7受体导致钾离子外流。此外,在胆管结扎(BDL)小鼠模型中,用半胱天冬酶-1抑制剂体内抑制NLRP3炎性小体可显著降低肝组织中成熟IL-1β水平,并改善肝纤维化。总之,过量的CDCA可能是一种内源性危险信号,可激活NLRP3炎性小体并在胆汁淤积期间引发肝脏炎症。我们的发现为通过靶向炎性小体激活来改善胆汁淤积性肝纤维化提供了机制基础。