Martín Verónica, Perales Celia, Fernández-Algar María, Dos Santos Helena G, Garrido Patricia, Pernas María, Parro Víctor, Moreno Miguel, García-Pérez Javier, Alcamí José, Torán José Luis, Abia David, Domingo Esteban, Briones Carlos
Centro de Biología Molecular 'Severo Ochoa' (CBMSO, CSIC-UAM). Campus de Cantoblanco, Madrid, Spain.
Centro de Investigación Biomédica en Red de enfermedades hepáticas y digestivas (CIBERehd), Spain.
PLoS One. 2016 Dec 13;11(12):e0166902. doi: 10.1371/journal.pone.0166902. eCollection 2016.
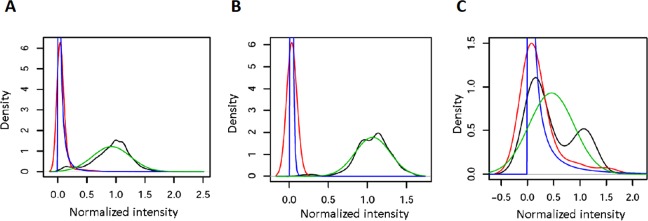
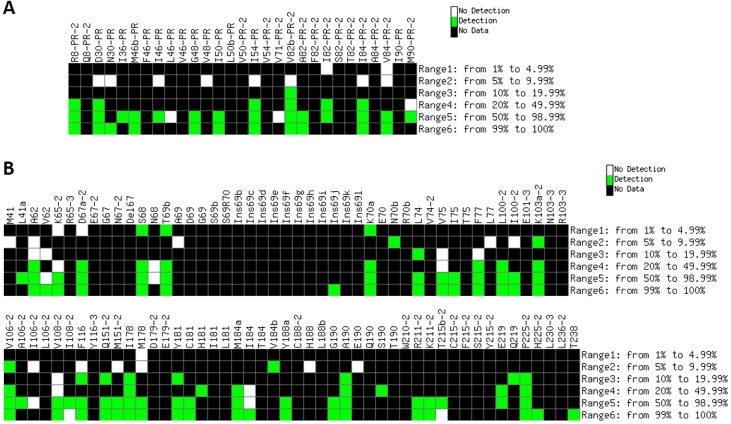
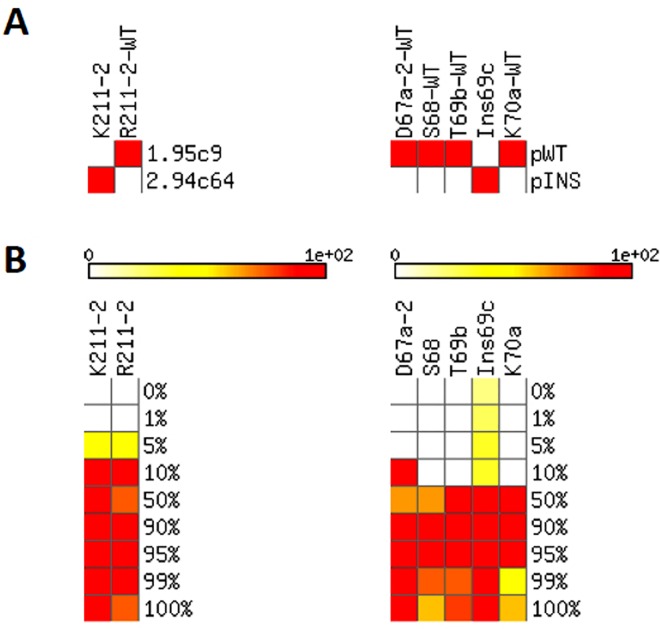
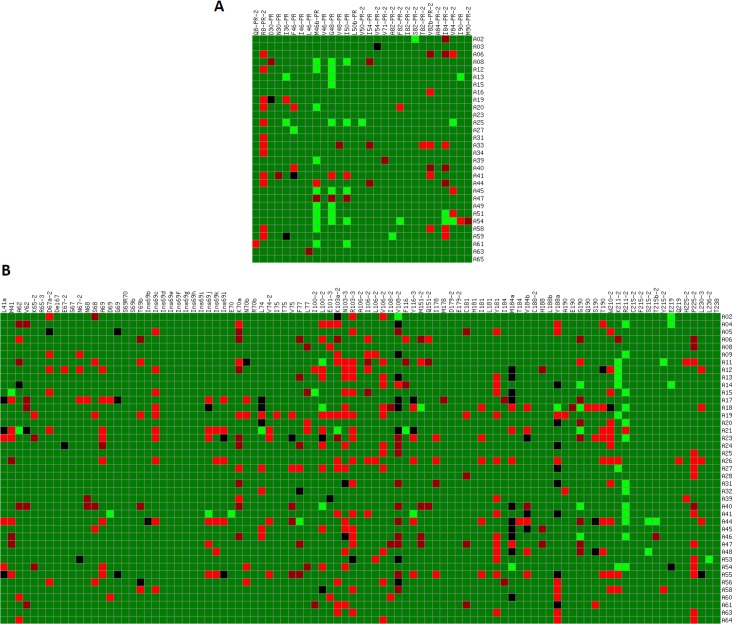
The response of human immunodeficiency virus type 1 (HIV-1) quasispecies to antiretroviral therapy is influenced by the ensemble of mutants that composes the evolving population. Low-abundance subpopulations within HIV-1 quasispecies may determine the viral response to the administered drug combinations. However, routine sequencing assays available to clinical laboratories do not recognize HIV-1 minority variants representing less than 25% of the population. Although several alternative and more sensitive genotyping techniques have been developed, including next-generation sequencing (NGS) methods, they are usually very time consuming, expensive and require highly trained personnel, thus becoming unrealistic approaches in daily clinical practice. Here we describe the development and testing of a HIV-1 genotyping DNA microarray that detects and quantifies, in majority and minority viral subpopulations, relevant mutations and amino acid insertions in 42 codons of the pol gene associated with drug- and multidrug-resistance to protease (PR) and reverse transcriptase (RT) inhibitors. A customized bioinformatics protocol has been implemented to analyze the microarray hybridization data by including a new normalization procedure and a stepwise filtering algorithm, which resulted in the highly accurate (96.33%) detection of positive/negative signals. This microarray has been tested with 57 subtype B HIV-1 clinical samples extracted from multi-treated patients, showing an overall identification of 95.53% and 89.24% of the queried PR and RT codons, respectively, and enough sensitivity to detect minority subpopulations representing as low as 5-10% of the total quasispecies. The developed genotyping platform represents an efficient diagnostic and prognostic tool useful to personalize antiviral treatments in clinical practice.
1型人类免疫缺陷病毒(HIV-1)准种对抗逆转录病毒疗法的反应受构成不断演变群体的突变体集合影响。HIV-1准种中的低丰度亚群可能决定病毒对所施用药物组合的反应。然而,临床实验室可用的常规测序检测无法识别占群体不到25%的HIV-1少数变异体。尽管已经开发了几种替代的、更灵敏的基因分型技术,包括下一代测序(NGS)方法,但它们通常非常耗时、昂贵且需要训练有素的人员,因此在日常临床实践中成为不切实际的方法。在此,我们描述了一种HIV-1基因分型DNA微阵列的开发和测试,该微阵列可检测和定量多数和少数病毒亚群中与蛋白酶(PR)和逆转录酶(RT)抑制剂的药物耐药和多药耐药相关的pol基因42个密码子中的相关突变和氨基酸插入。已经实施了定制的生物信息学方案来分析微阵列杂交数据,包括新的标准化程序和逐步过滤算法,这导致阳性/阴性信号的检测准确率高达96.33%。该微阵列已用从多次治疗患者中提取的57份B亚型HIV-1临床样本进行了测试,分别显示对所查询的PR和RT密码子的总体识别率为95.53%和89.24%,并且具有足够的灵敏度来检测占总准种低至5-10% 的少数亚群。所开发的基因分型平台代表了一种有效的诊断和预后工具,有助于在临床实践中实现抗病毒治疗的个性化。