Molecular Profiling Research Center for Drug Discovery (molprof), National Institute of Advanced Industrial Science and Technology (AIST), 2-3-26, Aomi, Koto-ku, Tokyo, 135-0064, Japan.
Technology Research Association for Next-Generation Natural Products Chemistry, 2-3-26, Aomi, Koto-ku, Tokyo, 135-0064, Japan.
Mol Inform. 2017 Jan;36(1-2). doi: 10.1002/minf.201600013. Epub 2016 Apr 29.
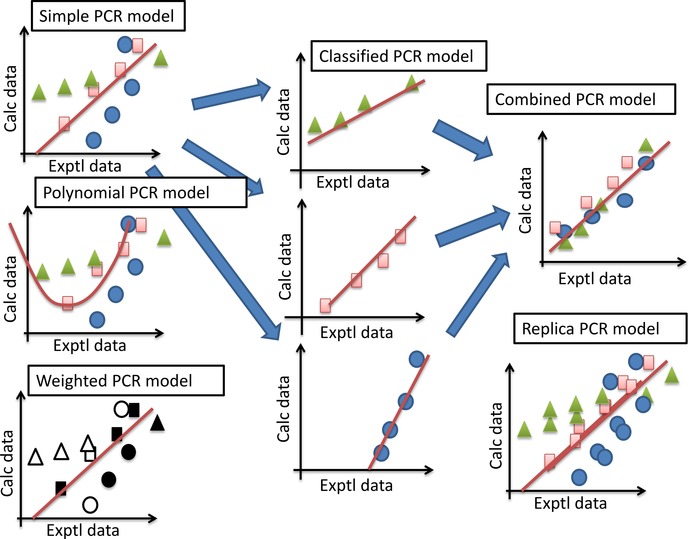
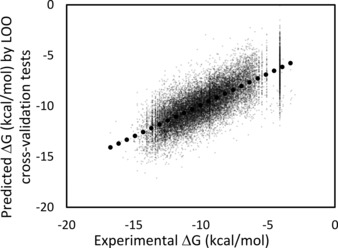
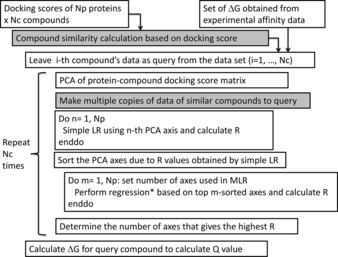
In order to improve docking score correction, we developed several structure-based quantitative structure activity relationship (QSAR) models by protein-drug docking simulations and applied these models to public affinity data. The prediction models used descriptor-based regression, and the compound descriptor was a set of docking scores against multiple (∼600) proteins including nontargets. The binding free energy that corresponded to the docking score was approximated by a weighted average of docking scores for multiple proteins, and we tried linear, weighted linear and polynomial regression models considering the compound similarities. In addition, we tried a combination of these regression models for individual data sets such as IC , K , and %inhibition values. The cross-validation results showed that the weighted linear model was more accurate than the simple linear regression model. Thus, the QSAR approaches based on the affinity data of public databases should improve docking scores.
为了提高对接评分的修正效果,我们通过蛋白-药物对接模拟开发了几个基于结构的定量构效关系(QSAR)模型,并将这些模型应用于公共亲和力数据。预测模型使用基于描述符的回归,化合物描述符是针对包括非靶点在内的多种(约 600 种)蛋白质的一组对接评分。与对接评分相对应的结合自由能通过对接评分的加权平均值来近似,我们考虑了化合物的相似性,尝试了线性、加权线性和多项式回归模型。此外,我们还尝试了将这些回归模型组合用于单个数据集,例如 IC 、 K 和 %抑制值。交叉验证结果表明,加权线性模型比简单线性回归模型更准确。因此,基于公共数据库的亲和力数据的 QSAR 方法应该可以提高对接评分。