Hoch Nicolas C, Hanzlikova Hana, Rulten Stuart L, Tétreault Martine, Komulainen Emilia, Ju Limei, Hornyak Peter, Zeng Zhihong, Gittens William, Rey Stephanie A, Staras Kevin, Mancini Grazia M S, McKinnon Peter J, Wang Zhao-Qi, Wagner Justin D, Yoon Grace, Caldecott Keith W
Genome Damage and Stability Centre, School of Life Sciences, University of Sussex, Falmer, Brighton BN1 9RH, UK.
CAPES Foundation, Ministry of Education of Brazil, Brasilia/DF 70040-020, Brazil.
Nature. 2017 Jan 5;541(7635):87-91. doi: 10.1038/nature20790. Epub 2016 Dec 21.
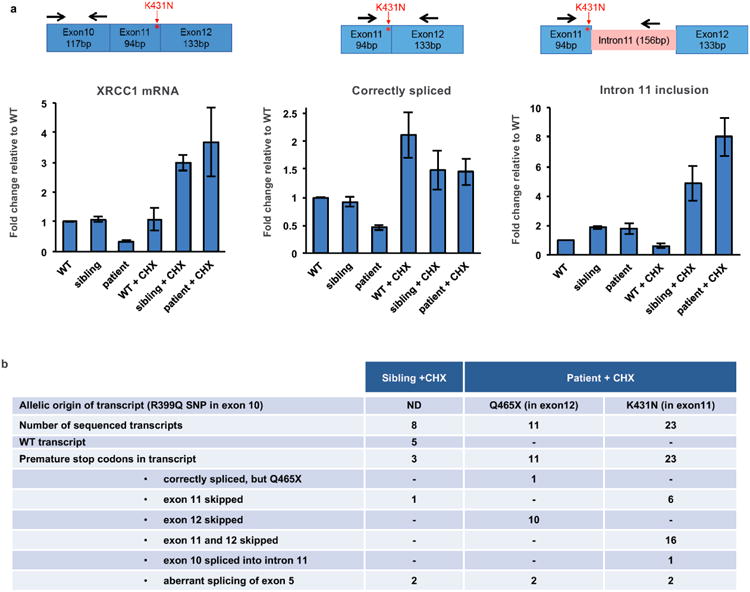
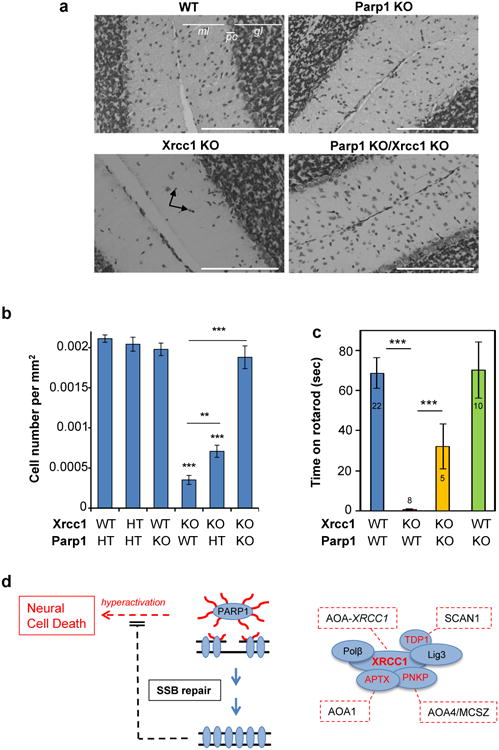
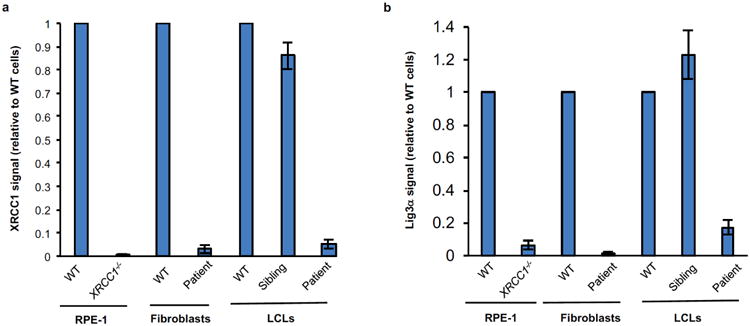
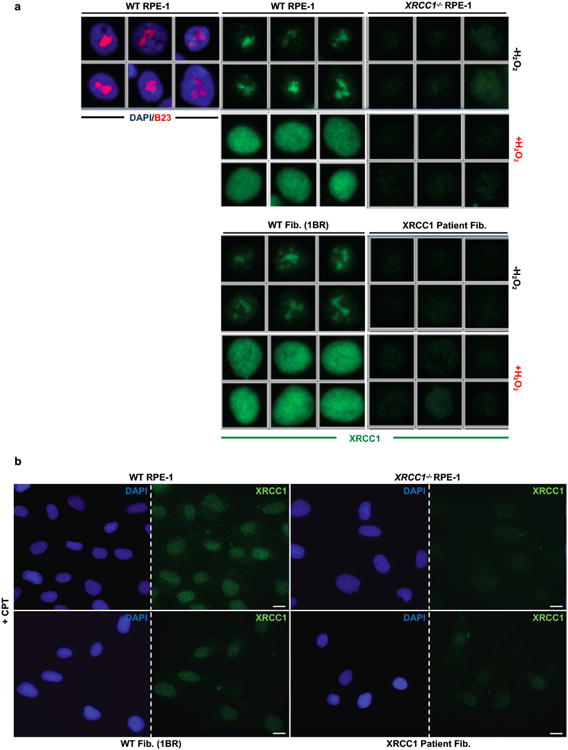
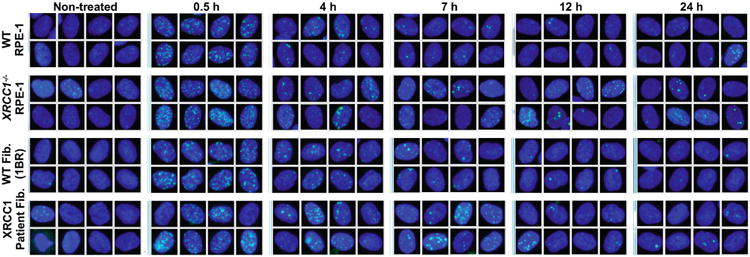
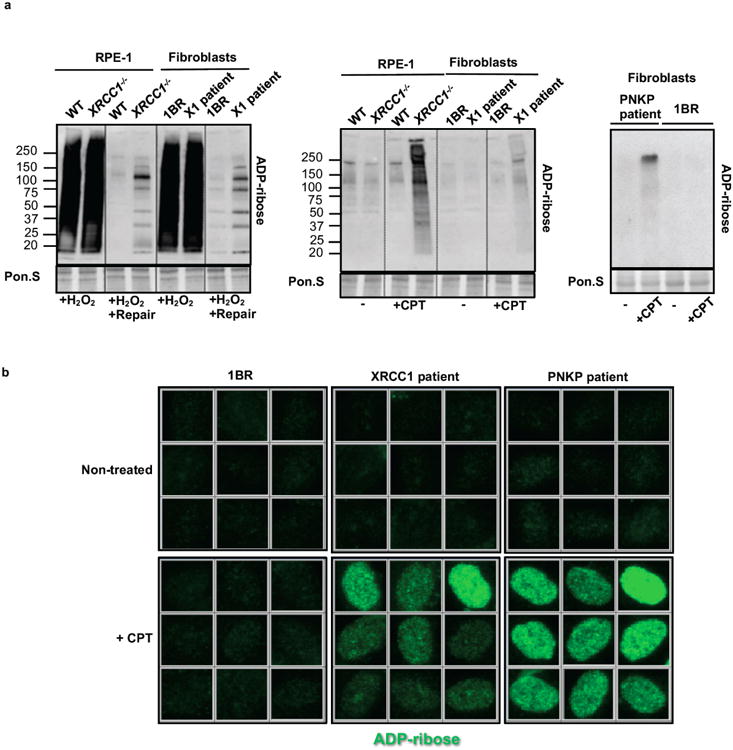
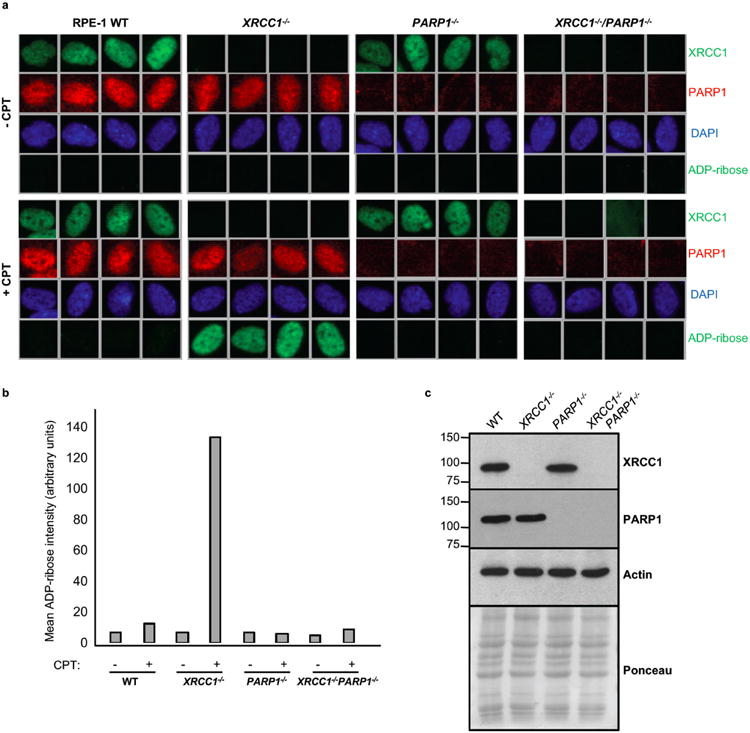
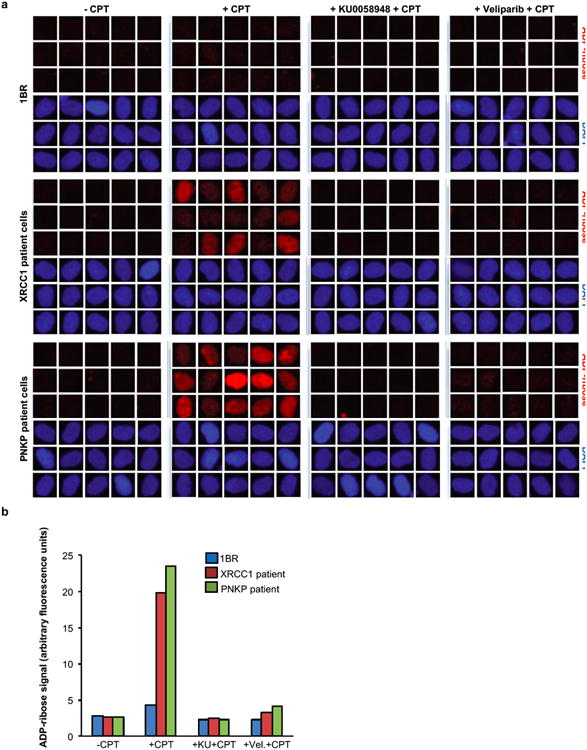
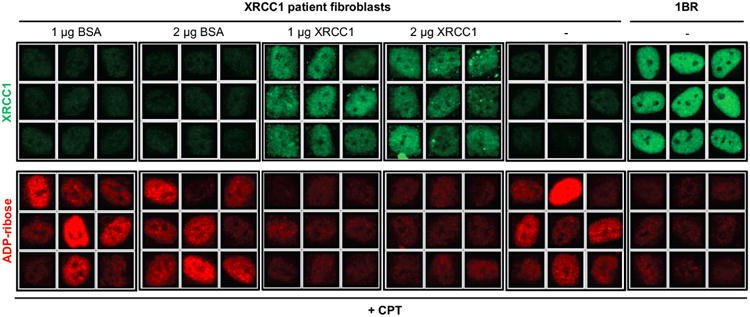
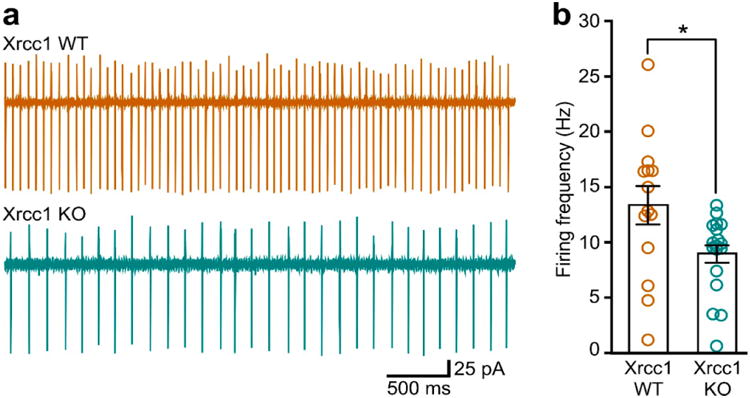
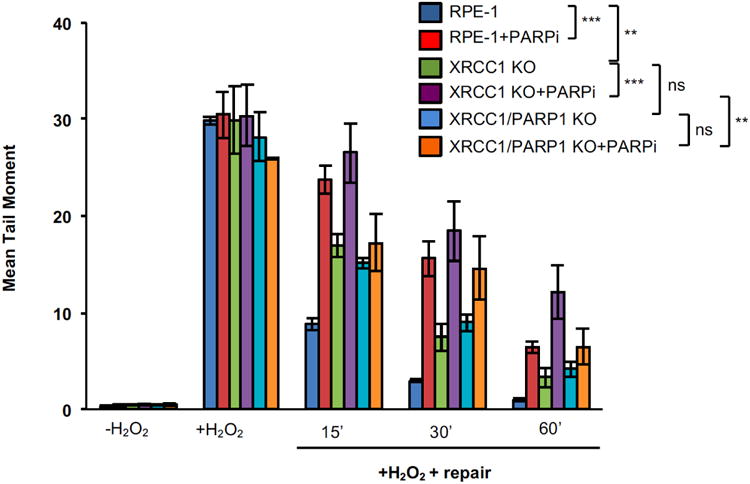
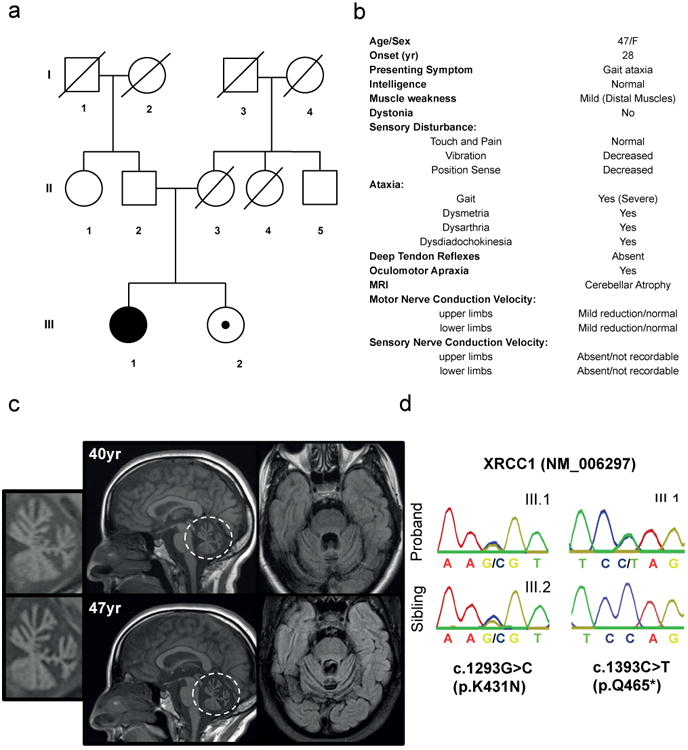
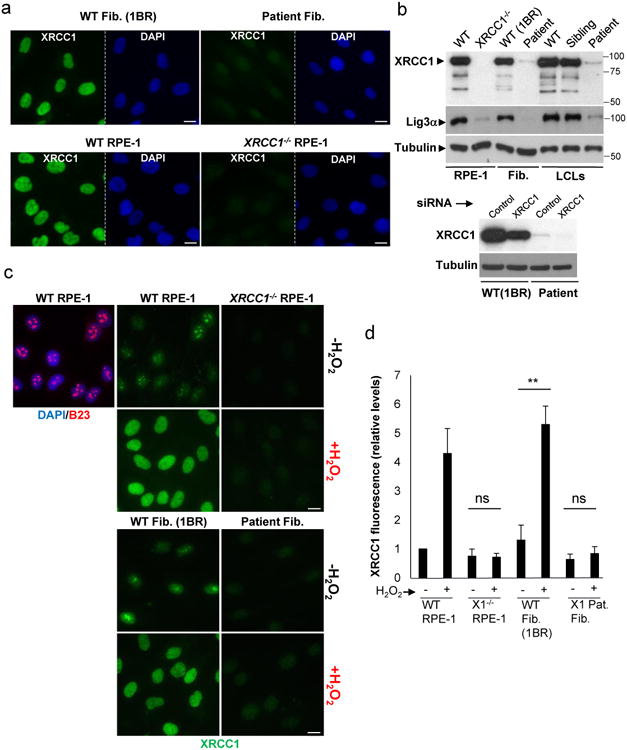
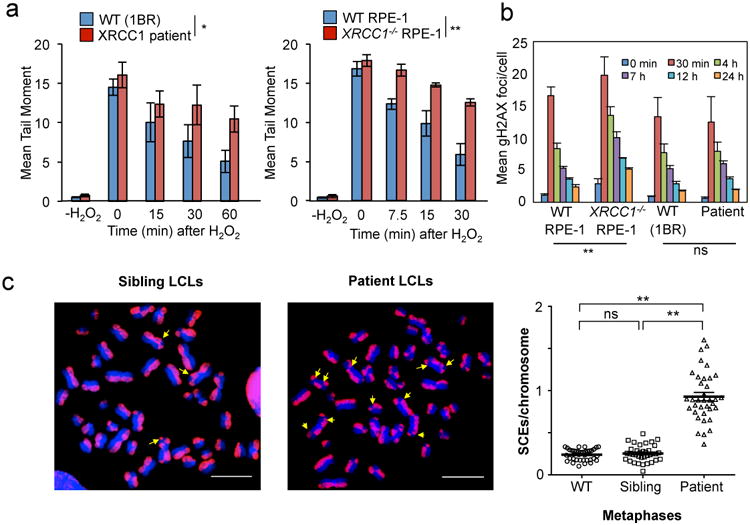
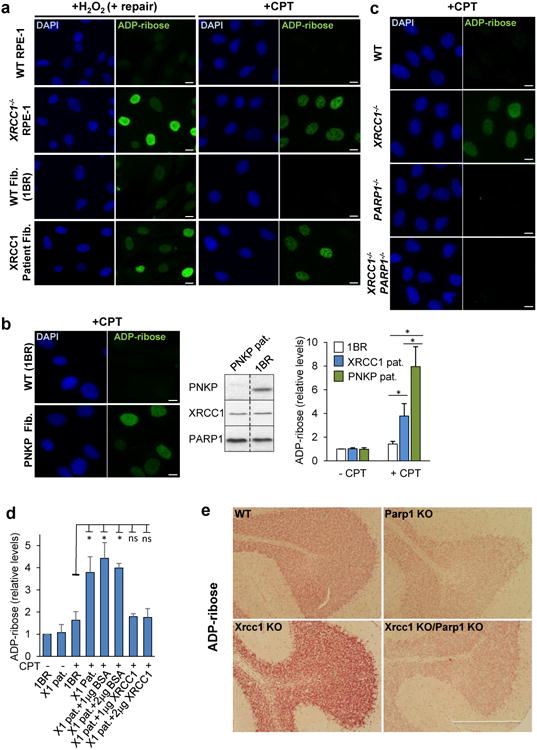
XRCC1 is a molecular scaffold protein that assembles multi-protein complexes involved in DNA single-strand break repair. Here we show that biallelic mutations in the human XRCC1 gene are associated with ocular motor apraxia, axonal neuropathy, and progressive cerebellar ataxia. Cells from a patient with mutations in XRCC1 exhibited not only reduced rates of single-strand break repair but also elevated levels of protein ADP-ribosylation. This latter phenotype is recapitulated in a related syndrome caused by mutations in the XRCC1 partner protein PNKP and implicates hyperactivation of poly(ADP-ribose) polymerase/s as a cause of cerebellar ataxia. Indeed, remarkably, genetic deletion of Parp1 rescued normal cerebellar ADP-ribose levels and reduced the loss of cerebellar neurons and ataxia in Xrcc1-defective mice, identifying a molecular mechanism by which endogenous single-strand breaks trigger neuropathology. Collectively, these data establish the importance of XRCC1 protein complexes for normal neurological function and identify PARP1 as a therapeutic target in DNA strand break repair-defective disease.
XRCC1是一种分子支架蛋白,可组装参与DNA单链断裂修复的多蛋白复合物。我们在此表明,人类XRCC1基因的双等位基因突变与眼球运动失用、轴索性神经病和进行性小脑共济失调相关。来自一名XRCC1基因突变患者的细胞不仅单链断裂修复率降低,而且蛋白质ADP-核糖基化水平升高。后一种表型在由XRCC1伴侣蛋白PNKP突变引起的相关综合征中也有体现,这表明聚(ADP-核糖)聚合酶的过度激活是小脑共济失调的一个原因。实际上,值得注意的是,Parp1的基因缺失挽救了正常的小脑ADP-核糖水平,并减少了Xrcc1缺陷小鼠中小脑神经元的损失和共济失调,从而确定了内源性单链断裂引发神经病理学的分子机制。总体而言,这些数据确立了XRCC1蛋白复合物对正常神经功能的重要性,并将PARP1确定为DNA链断裂修复缺陷疾病的治疗靶点。