Farrar Michelle A, Park Susanna B, Vucic Steve, Carey Kate A, Turner Bradley J, Gillingwater Thomas H, Swoboda Kathryn J, Kiernan Matthew C
Discipline of Paediatrics, School of Women's and Children's Health, UNSW Medicine, The University of New South Wales, Sydney, Australia.
Brain & Mind Centre and Sydney Medical School, University of Sydney, Sydney, Australia.
Ann Neurol. 2017 Mar;81(3):355-368. doi: 10.1002/ana.24864. Epub 2017 Feb 17.
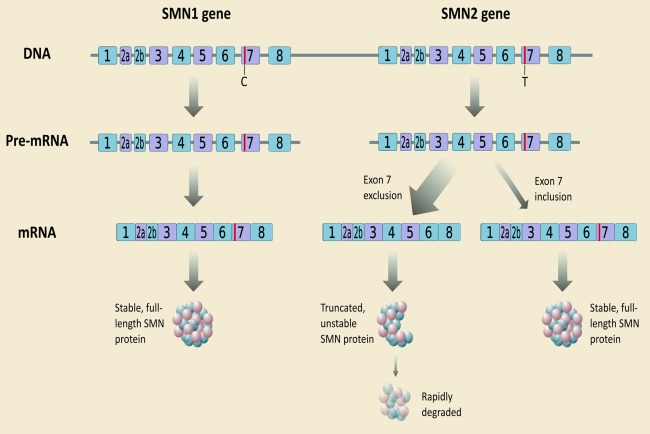
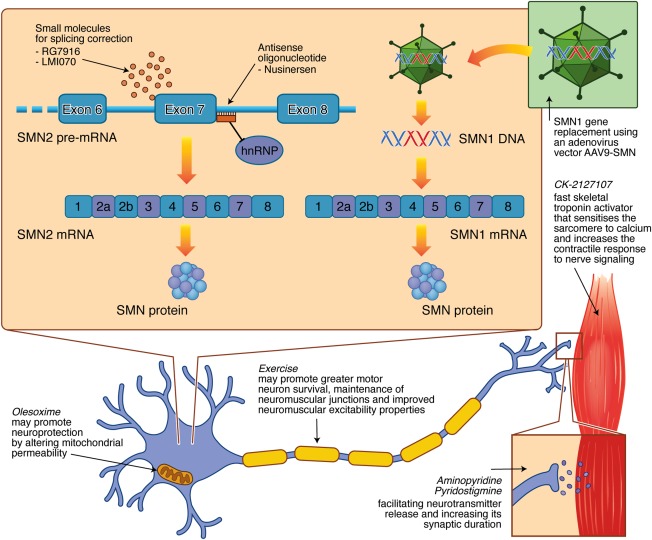
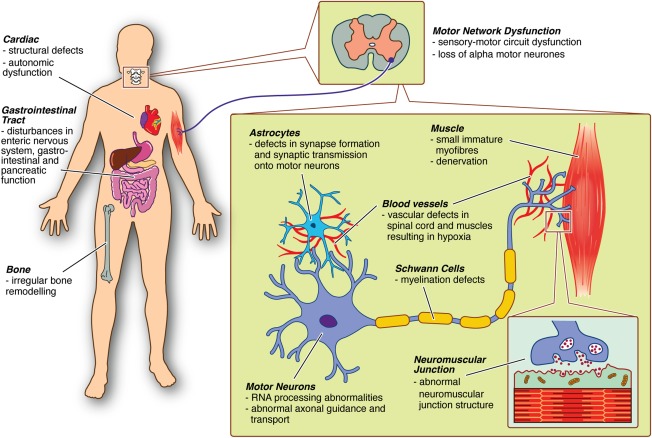
Spinal muscular atrophy (SMA) is a hereditary neurodegenerative disease with severity ranging from progressive infantile paralysis and premature death (type I) to limited motor neuron loss and normal life expectancy (type IV). Without disease-modifying therapies, the impact is profound for patients and their families. Improved understanding of the molecular basis of SMA, disease pathogenesis, natural history, and recognition of the impact of standardized care on outcomes has yielded progress toward the development of novel therapeutic strategies and are summarized. Therapeutic strategies in the pipeline are appraised, ranging from SMN1 gene replacement to modulation of SMN2 encoded transcripts, to neuroprotection, to an expanding repertoire of peripheral targets, including muscle. With the advent of preliminary trial data, it can be reasonably anticipated that the SMA treatment landscape will transform significantly. Advancement in presymptomatic diagnosis and screening programs will be critical, with pilot newborn screening studies underway to facilitate preclinical diagnosis. The development of disease-modifying therapies will necessitate monitoring programs to determine the long-term impact, careful evaluation of combined treatments, and further acceleration of improvements in supportive care. In advance of upcoming clinical trial results, we consider the challenges and controversies related to the implementation of novel therapies for all patients and set the scene as the field prepares to enter an era of novel therapies. Ann Neurol 2017;81:355-368.
脊髓性肌萎缩症(SMA)是一种遗传性神经退行性疾病,严重程度从进行性婴儿麻痹和过早死亡(I型)到有限的运动神经元丧失和正常预期寿命(IV型)不等。如果没有疾病改善疗法,对患者及其家庭的影响将是深远的。对SMA分子基础、疾病发病机制、自然史的进一步了解,以及对标准化护理对预后影响的认识,为新型治疗策略的开发带来了进展,并进行了总结。正在评估一系列治疗策略,从SMN1基因替代到对SMN2编码转录本的调控,再到神经保护,以及包括肌肉在内的越来越多的外周靶点。随着初步试验数据的出现,可以合理预期SMA的治疗格局将发生显著变化。症状前诊断和筛查项目的进展至关重要,目前正在进行试点新生儿筛查研究以促进临床前诊断。疾病改善疗法的发展将需要监测项目来确定长期影响,仔细评估联合治疗,并进一步加快支持性护理的改善。在即将公布临床试验结果之前,我们考虑了与为所有患者实施新型疗法相关的挑战和争议,并在该领域准备进入新型疗法时代之际做好铺垫。《神经病学纪事》2017年;81:355 - 368。