Safari-Alighiarloo Nahid, Rezaei-Tavirani Mostafa, Taghizadeh Mohammad, Tabatabaei Seyyed Mohammad, Namaki Saeed
Proteomics Research Center, Department of Basic Science, Faculty of Paramedical Sciences, Shahid Beheshti University of Medical Sciences , Tehran , Iran.
Bioinformatics Department, Institute of Biochemistry and Biophysics, Tehran University , Tehran , Iran.
PeerJ. 2016 Dec 22;4:e2775. doi: 10.7717/peerj.2775. eCollection 2016.
The involvement of multiple genes and missing heritability, which are dominant in complex diseases such as multiple sclerosis (MS), entail using network biology to better elucidate their molecular basis and genetic factors. We therefore aimed to integrate interactome (protein-protein interaction (PPI)) and transcriptomes data to construct and analyze PPI networks for MS disease.
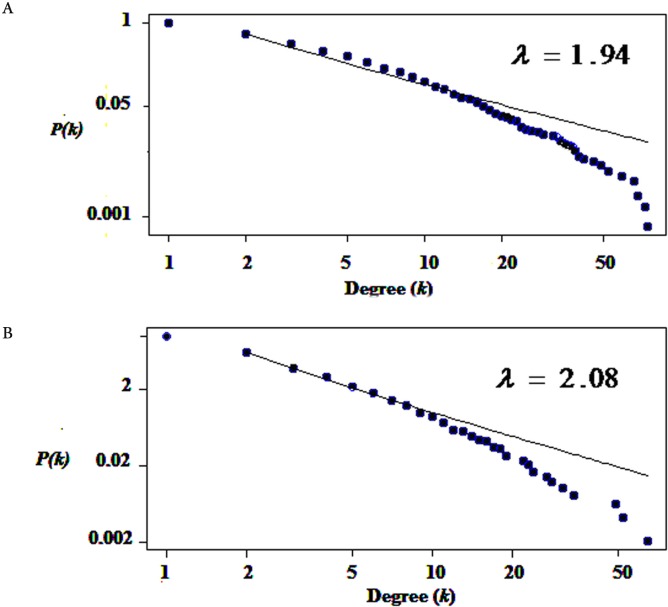
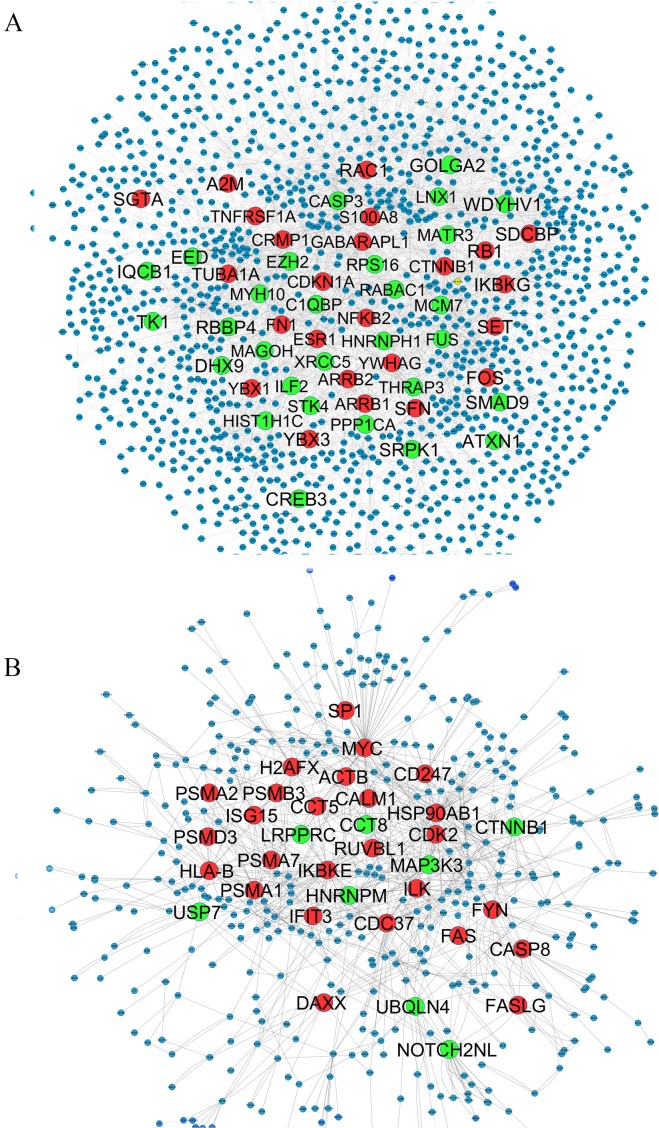
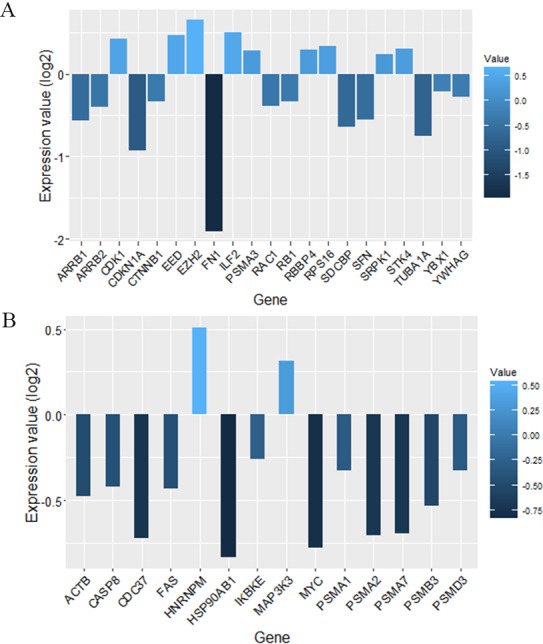
Gene expression profiles in paired cerebrospinal fluid (CSF) and peripheral blood mononuclear cells (PBMCs) samples from MS patients, sampled in relapse or remission and controls, were analyzed. Differentially expressed genes which determined only in CSF (MS control) and PBMCs (relapse remission) separately integrated with PPI data to construct the Query-Query PPI (QQPPI) networks. The networks were further analyzed to investigate more central genes, functional modules and complexes involved in MS progression.
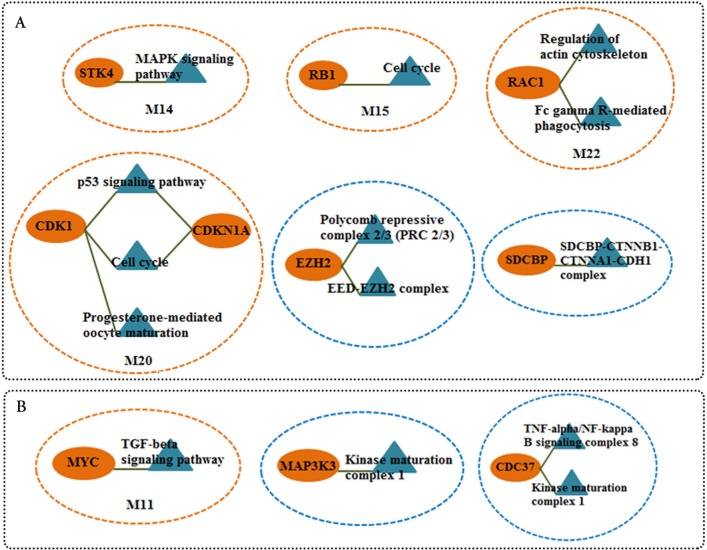
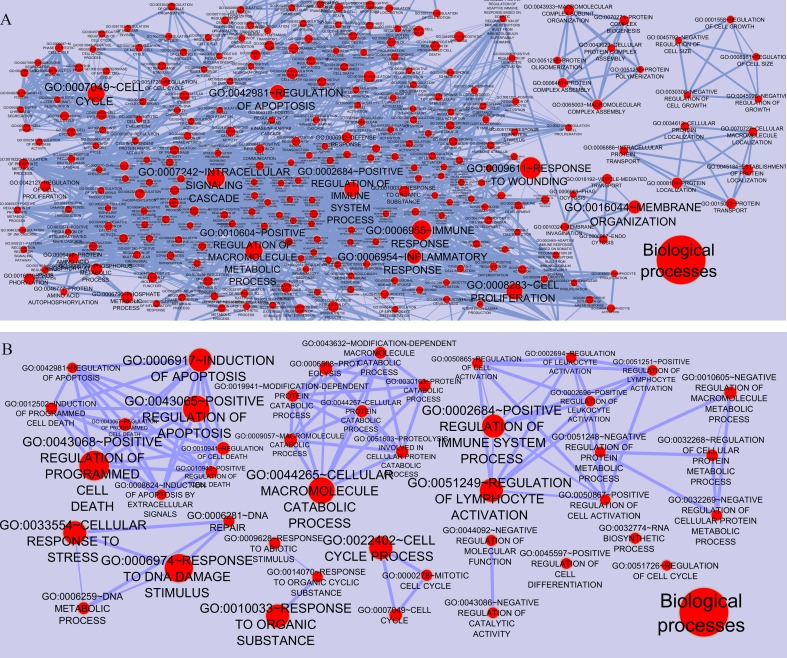
The networks were analyzed and high centrality genes were identified. Exploration of functional modules and complexes showed that the majority of high centrality genes incorporated in biological pathways driving MS pathogenesis. Proteasome and spliceosome were also noticeable in enriched pathways in PBMCs (relapse remission) which were identified by both modularity and clique analyses. Finally, STK4, RB1, CDKN1A, CDK1, RAC1, EZH2, SDCBP genes in CSF (MS control) and CDC37, MAP3K3, MYC genes in PBMCs (relapse remission) were identified as potential candidate genes for MS, which were the more central genes involved in biological pathways.
This study showed that network-based analysis could explicate the complex interplay between biological processes underlying MS. Furthermore, an experimental validation of candidate genes can lead to identification of potential therapeutic targets.
多种基因的参与和遗传力缺失在诸如多发性硬化症(MS)等复杂疾病中占主导地位,这就需要利用网络生物学来更好地阐明其分子基础和遗传因素。因此,我们旨在整合相互作用组(蛋白质 - 蛋白质相互作用(PPI))和转录组数据,以构建和分析MS疾病的PPI网络。
分析了来自MS患者、处于复发或缓解期的患者以及对照的配对脑脊液(CSF)和外周血单核细胞(PBMC)样本中的基因表达谱。仅在CSF(MS与对照)和PBMC(复发与缓解)中分别确定的差异表达基因与PPI数据整合,以构建查询 - 查询PPI(QQPPI)网络。对这些网络进行进一步分析,以研究更多参与MS进展的核心基因、功能模块和复合物。
对网络进行了分析并鉴定出了高中心性基因。对功能模块和复合物的探索表明,大多数高中心性基因纳入了驱动MS发病机制的生物途径。蛋白酶体和剪接体在PBMC(复发与缓解)的富集途径中也很明显,这是通过模块性和团分析确定的。最后,CSF(MS与对照)中的STK4、RB1、CDKN1A、CDK1、RAC1、EZH2、SDCBP基因以及PBMC(复发与缓解)中的CDC37、MAP3K3、MYC基因被鉴定为MS的潜在候选基因,它们是参与生物途径的更核心基因。
本研究表明,基于网络的分析可以阐明MS潜在生物过程之间的复杂相互作用。此外,对候选基因的实验验证可能会导致潜在治疗靶点的识别。