Rhee Ji Young, Song Jae Hoon, Ko Kwan Soo
Division of Infectious Diseases, Department of Medicine, Dankook University, Cheonan, Korea.
Division of Infectious Diseases, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
Infect Chemother. 2016 Dec;48(4):285-293. doi: 10.3947/ic.2016.48.4.285.
Stenotrophomonas maltophilia is one of several opportunistic pathogens of growing significance. Several studies on the molecular epidemiology of S. maltophilia have shown clinical isolates to be genetically diverse.
A total of 121 clinical isolates tentatively identified as S. malophilia from seven tertiary-care hospitals in Korea from 2007 to 2011 were included. Species and groups were identified using partial gyrB gene sequences and antimicrobial susceptibility testing was performed using a broth microdilution method. Multi locus variable number of tandem repeat analysis (MLVA) surveys are used for subtyping.
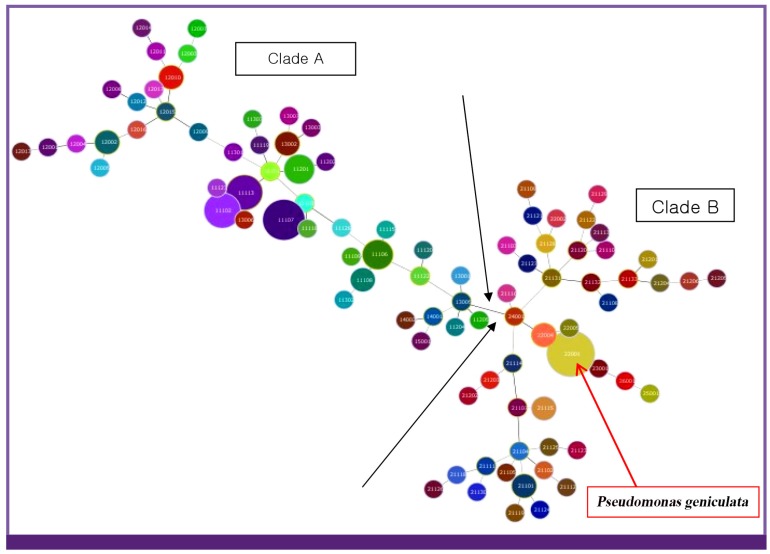
Based on partial gyrB gene sequences, 118 isolates were identified as belonging to the S. maltophilia complex. For all S. maltophilia isolates, the resistance rates to trimethoprime-sulfamethoxazole (TMP/SMX) and levofloxacin were the highest (both, 30.5%). Resistance rate to ceftazidime was 28.0%. 11.0% and 11.9% of 118 S. maltophilia isolates displayed resistance to piperacillin/tazobactam and tigecycline, respectively. Clade 1 and Clade 2 were definitely distinguished from the data of MLVA with amplification of loci. All 118 isolates were classified into several clusters as its identification.
Because of high resistance rates to TMP/SMX and levofloxacin, the clinical laboratory department should consider providing the data about other antimicrobial agents and treatment of S. maltophilia infections with a combination of antimicrobials can be considered in the current practice. The MLVA evaluated in this study provides a fast, portable, relatively low cost genotyping method that can be employed in genotypic linkage or transmission networks comparing to analysis of the gyrB gene.
嗜麦芽窄食单胞菌是几种具有越来越重要意义的机会性病原体之一。多项关于嗜麦芽窄食单胞菌分子流行病学的研究表明,临床分离株在基因上具有多样性。
纳入了2007年至2011年从韩国七家三级医疗医院初步鉴定为嗜麦芽窄食单胞菌的121株临床分离株。使用部分gyrB基因序列鉴定菌种和菌群,并采用肉汤微量稀释法进行药敏试验。多基因座可变数目串联重复分析(MLVA)调查用于亚型分析。
基于部分gyrB基因序列,118株分离株被鉴定为属于嗜麦芽窄食单胞菌复合体。对于所有嗜麦芽窄食单胞菌分离株,对复方新诺明(TMP/SMX)和左氧氟沙星的耐药率最高(均为30.5%)。对头孢他啶的耐药率为28.0%。118株嗜麦芽窄食单胞菌分离株中,分别有11.0%和11.9%对哌拉西林/他唑巴坦和替加环素耐药。通过位点扩增的MLVA数据明确区分了进化枝1和进化枝2。所有118株分离株根据其鉴定结果被分为几个簇。
由于对TMP/SMX和左氧氟沙星的耐药率较高,临床实验室部门应考虑提供其他抗菌药物的数据,目前实践中可考虑联合使用抗菌药物治疗嗜麦芽窄食单胞菌感染。与gyrB基因分析相比,本研究中评估的MLVA提供了一种快速、便携、成本相对较低的基因分型方法,可用于基因连锁或传播网络分析。