Taghipour Shirin, Zarrineh Peyman, Ganjtabesh Mohammad, Nowzari-Dalini Abbas
Department of Computer Science, School of Mathematics, Statistics, and Computer Science, University of Tehran, P.O.Box: 14155-6455, Tehran, Iran.
BMC Bioinformatics. 2017 Jan 3;18(1):10. doi: 10.1186/s12859-016-1422-x.
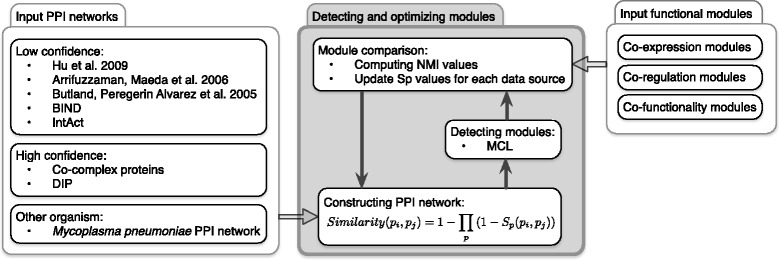
Although different protein-protein physical interaction (PPI) datasets exist for Escherichia coli, no common methodology exists to integrate these datasets and extract reliable modules reflecting the existing biological process and protein complexes. Naïve Bayesian formula is the highly accepted method to integrate different PPI datasets into a single weighted PPI network, but detecting proper weights in such network is still a major problem.
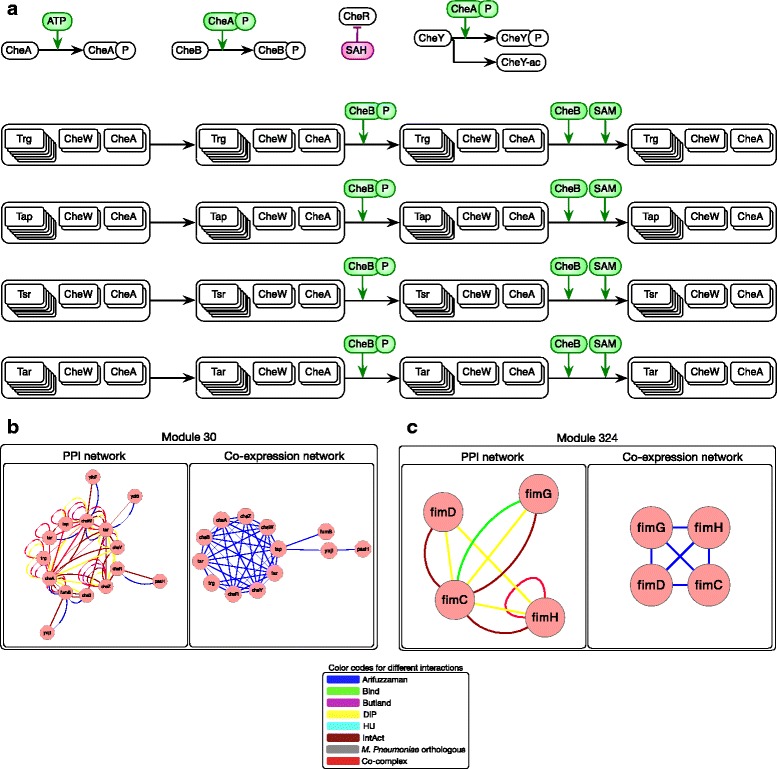
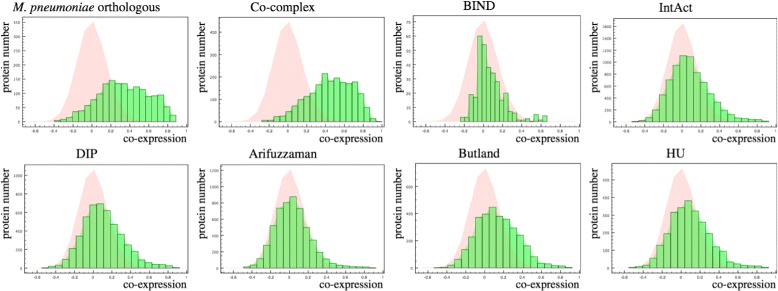
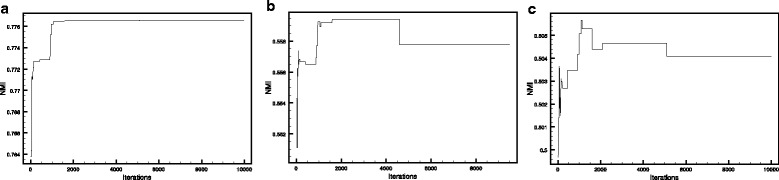
In this paper, we proposed a new methodology to integrate various physical PPI datasets into a single weighted PPI network in a way that the detected modules in PPI network exhibit the highest similarity to available functional modules. We used the co-expression modules as functional modules, and we shown that direct functional modules detected from Gene Ontology terms could be used as an alternative dataset. After running this integrating methodology over six different physical PPI datasets, orthologous high-confidence interactions from a related organism and two AP-MS PPI datasets gained high weights in the integrated networks, while the weights for one AP-MS PPI dataset and two other datasets derived from public databases have converged to zero. The majority of detected modules shaped around one or few hub protein(s). Still, a large number of highly interacting protein modules were detected which are functionally relevant and are likely to construct protein complexes.
We provided a new high confidence protein complex prediction method supported by functional studies and literature mining.
尽管存在不同的大肠杆菌蛋白质-蛋白质物理相互作用(PPI)数据集,但不存在整合这些数据集并提取反映现有生物过程和蛋白质复合物的可靠模块的通用方法。朴素贝叶斯公式是将不同PPI数据集整合到单个加权PPI网络中被广泛接受的方法,但在此类网络中检测合适的权重仍然是一个主要问题。
在本文中,我们提出了一种新方法,将各种物理PPI数据集整合到单个加权PPI网络中,使得PPI网络中检测到的模块与可用功能模块具有最高的相似性。我们将共表达模块用作功能模块,并且表明从基因本体术语中检测到的直接功能模块可以用作替代数据集。在六个不同的物理PPI数据集上运行这种整合方法后,来自相关生物体的直系同源高置信度相互作用以及两个亲和纯化-质谱(AP-MS)PPI数据集在整合网络中获得了高权重,而一个AP-MS PPI数据集和另外两个来自公共数据库的数据集的权重已收敛到零。大多数检测到的模块围绕一个或几个枢纽蛋白形成。尽管如此,仍检测到大量高度相互作用的蛋白质模块,它们在功能上相关并且可能构建蛋白质复合物。
我们提供了一种由功能研究和文献挖掘支持的新的高置信度蛋白质复合物预测方法。