Rodríguez-Martín Bernardo, Palumbo Emilio, Marco-Sola Santiago, Griebel Thasso, Ribeca Paolo, Alonso Graciela, Rastrojo Alberto, Aguado Begoña, Guigó Roderic, Djebali Sarah
Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, Dr. Aiguader 88, Barcelona, 08003, Spain.
Universitat Pompeu Fabra (UPF), Barcelona, Spain.
BMC Genomics. 2017 Jan 3;18(1):7. doi: 10.1186/s12864-016-3404-9.
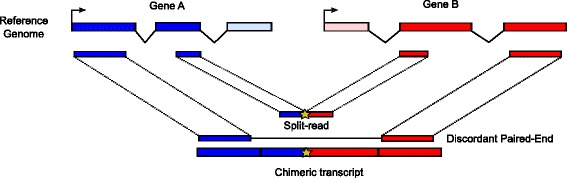
Chimeric transcripts are commonly defined as transcripts linking two or more different genes in the genome, and can be explained by various biological mechanisms such as genomic rearrangement, read-through or trans-splicing, but also by technical or biological artefacts. Several studies have shown their importance in cancer, cell pluripotency and motility. Many programs have recently been developed to identify chimeras from Illumina RNA-seq data (mostly fusion genes in cancer). However outputs of different programs on the same dataset can be widely inconsistent, and tend to include many false positives. Other issues relate to simulated datasets restricted to fusion genes, real datasets with limited numbers of validated cases, result inconsistencies between simulated and real datasets, and gene rather than junction level assessment.
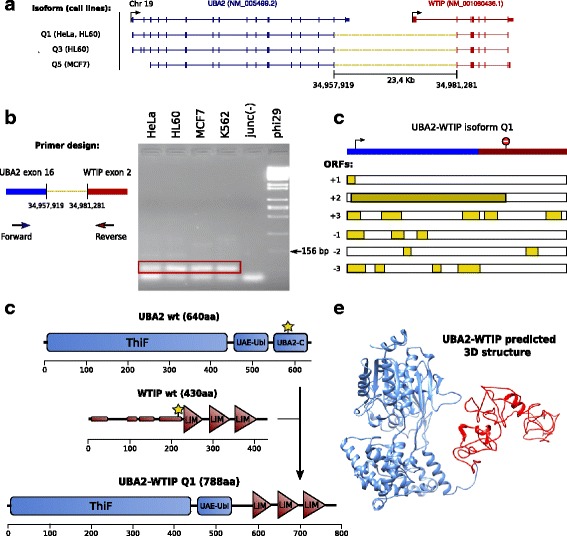
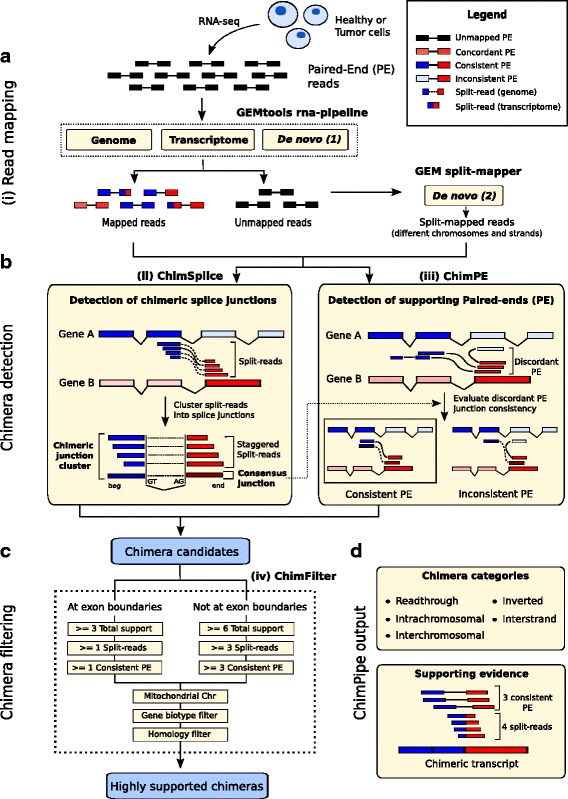
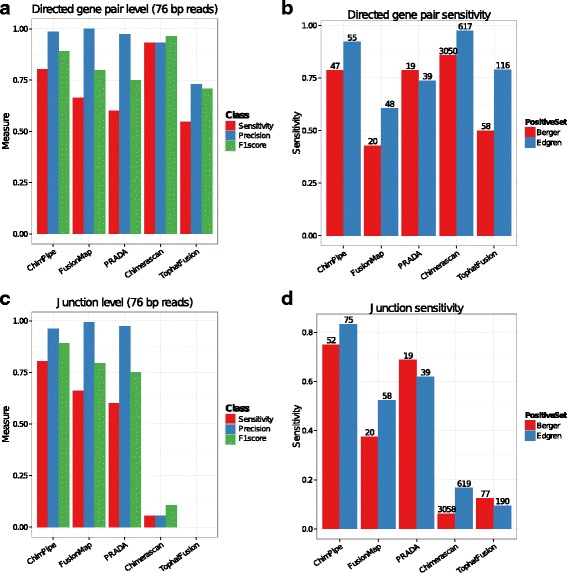
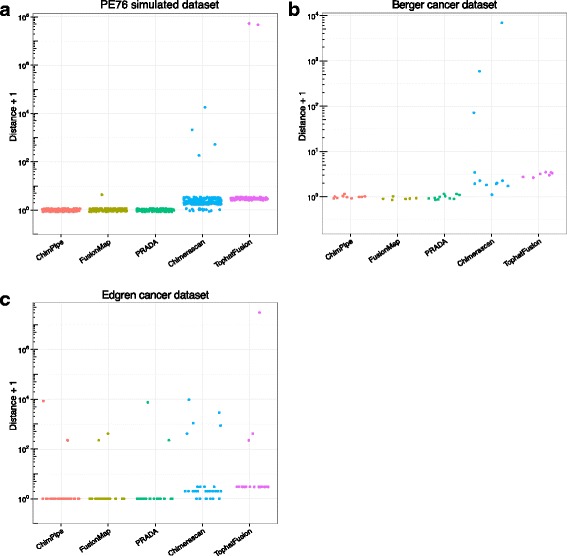
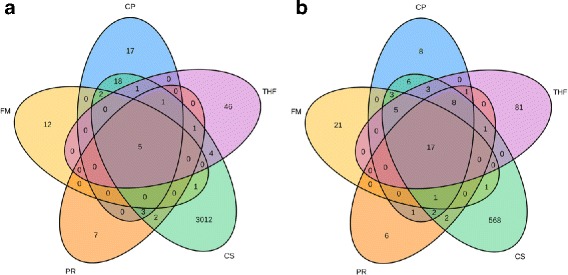
Here we present ChimPipe, a modular and easy-to-use method to reliably identify fusion genes and transcription-induced chimeras from paired-end Illumina RNA-seq data. We have also produced realistic simulated datasets for three different read lengths, and enhanced two gold-standard cancer datasets by associating exact junction points to validated gene fusions. Benchmarking ChimPipe together with four other state-of-the-art tools on this data showed ChimPipe to be the top program at identifying exact junction coordinates for both kinds of datasets, and the one showing the best trade-off between sensitivity and precision. Applied to 106 ENCODE human RNA-seq datasets, ChimPipe identified 137 high confidence chimeras connecting the protein coding sequence of their parent genes. In subsequent experiments, three out of four predicted chimeras, two of which recurrently expressed in a large majority of the samples, could be validated. Cloning and sequencing of the three cases revealed several new chimeric transcript structures, 3 of which with the potential to encode a chimeric protein for which we hypothesized a new role. Applying ChimPipe to human and mouse ENCODE RNA-seq data led to the identification of 131 recurrent chimeras common to both species, and therefore potentially conserved.
ChimPipe combines discordant paired-end reads and split-reads to detect any kind of chimeras, including those originating from polymerase read-through, and shows an excellent trade-off between sensitivity and precision. The chimeras found by ChimPipe can be validated in-vitro with high accuracy.
嵌合转录本通常被定义为连接基因组中两个或多个不同基因的转录本,其产生可由多种生物学机制解释,如基因组重排、通读或反式剪接,也可能是技术或生物学假象导致的。多项研究表明它们在癌症、细胞多能性和运动性方面具有重要意义。最近已经开发了许多程序来从Illumina RNA测序数据中识别嵌合体(主要是癌症中的融合基因)。然而,不同程序对同一数据集的输出可能存在很大差异,并且往往包含许多假阳性结果。其他问题包括仅限于融合基因的模拟数据集、经过验证的病例数量有限的真实数据集、模拟数据集和真实数据集之间的结果不一致,以及基因水平而非连接点水平的评估。
在此,我们介绍ChimPipe,一种模块化且易于使用的方法,可从双末端Illumina RNA测序数据中可靠地识别融合基因和转录诱导的嵌合体。我们还针对三种不同的读长生成了逼真的模拟数据集,并通过将精确的连接点与经过验证的基因融合相关联,增强了两个金标准癌症数据集。将ChimPipe与其他四个最先进的工具在此数据上进行基准测试,结果表明ChimPipe是在识别两种数据集的精确连接坐标方面表现最佳的程序,并且在灵敏度和精度之间表现出最佳的权衡。应用于106个ENCODE人类RNA测序数据集时,ChimPipe识别出137个连接其亲本基因蛋白质编码序列的高可信度嵌合体。在后续实验中,四个预测的嵌合体中有三个可以得到验证,其中两个在大多数样本中反复表达。对这三个案例进行克隆和测序揭示了几种新的嵌合转录本结构,其中3种有可能编码嵌合蛋白,我们对其功能提出了新的假设。将ChimPipe应用于人类和小鼠的ENCODE RNA测序数据,导致识别出131个两种物种共有的反复出现的嵌合体,因此可能是保守的。
ChimPipe结合了不一致的双末端读段和分裂读段来检测任何类型的嵌合体,包括那些源自聚合酶通读产生的嵌合体,并且在灵敏度和精度之间表现出出色的权衡。ChimPipe发现的嵌合体可以在体外以高精度进行验证。