Gupta Sudheer, Sharma Ashok K, Shastri Vibhuti, Madhu Midhun K, Sharma Vineet K
Metagenomics and Systems Biology Group, Department of Biological Sciences, Indian Institute of Science Education and Research Bhopal, Bhopal, India.
J Transl Med. 2017 Jan 6;15(1):7. doi: 10.1186/s12967-016-1103-6.
The current therapy for inflammatory and autoimmune disorders involves the use of nonspecific anti-inflammatory drugs and other immunosuppressant, which are often accompanied with potential side effects. As an alternative therapy, anti-inflammatory peptides are recently being exploited as anti-inflammatory agents for treatment of various inflammatory diseases such as Alzheimer's disease and rheumatoid arthritis. Thus, understanding the correlation between amino acid sequence and its potential anti-inflammatory property is of great importance for the discovery of novel and efficient anti-inflammatory peptide-based therapeutics.
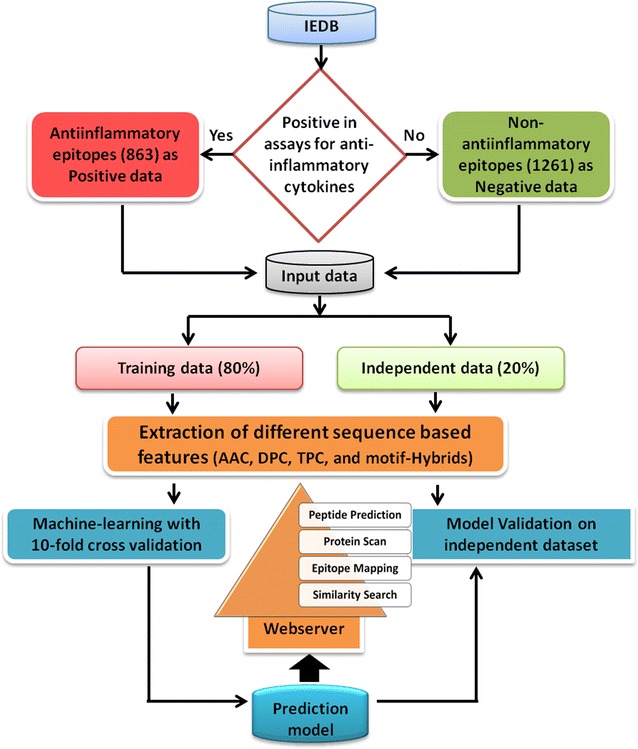
In this study, we have developed a prediction tool for the classification of peptides as anti-inflammatory epitopes or non anti-inflammatory epitopes. The training was performed using experimentally validated epitopes obtained from Immune epitope database and analysis resource database. Different sequence-based features and their hybrids with motif information were employed for development of support vector machine-based machine learning models. Similarly, machine learning models were also constructed using random forest.
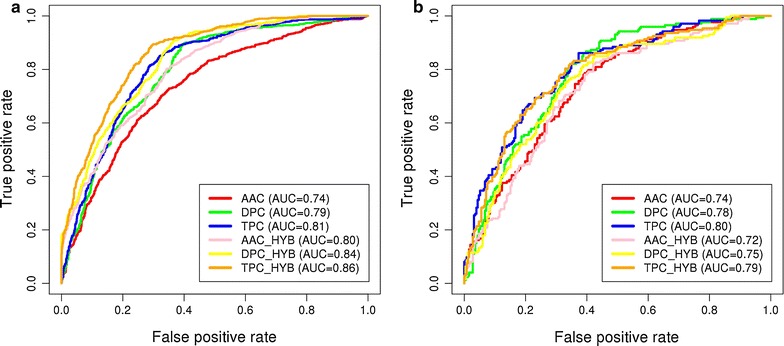
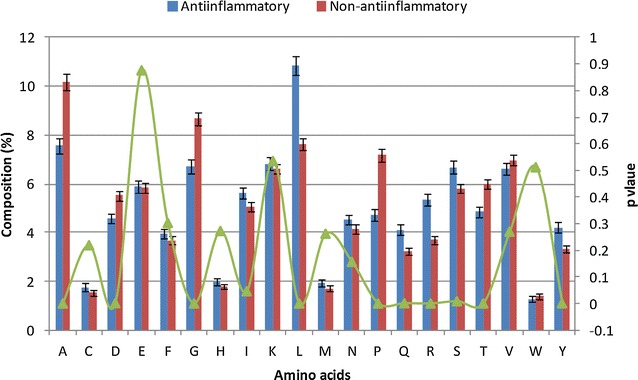
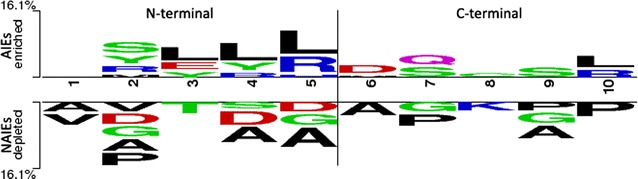
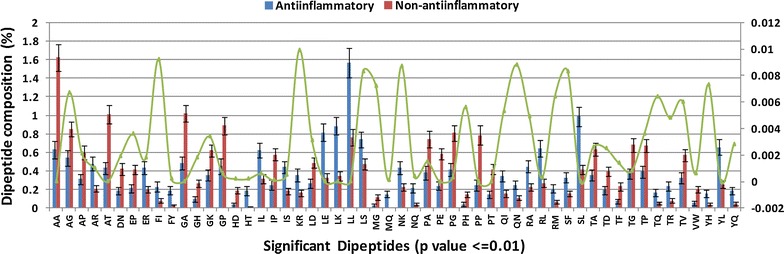
The composition and terminal residue conservation analysis of peptides revealed the dominance of leucine, serine, tyrosine and arginine residues in anti-inflammatory epitopes as compared to non anti-inflammatory epitopes. Similarly, the anti-inflammatory epitopes specific motifs were found to be rich in hydrophobic and polar residues. The hybrid of tripeptide composition-based support vector machine model and motif yielded the best performance on 10-fold cross validation with an accuracy of 78.1% and MCC of 0.58. The same displayed an accuracy of 72% and MCC of 0.45 on validation dataset, rejecting any possibility of over-fitting. The tripeptide composition-based random forest model displayed an accuracy of 0.8 and MCC of 0.59 on 10-fold cross validation, however, the accuracy (0.68) and MCC (0.31) was lower as compared to support vector machine model on validation dataset. Thus, the support vector machine model is implemented as the default model and an additional option of using the random forest model is provided.
The prediction models along with tools for epitope mapping and similarity search have been provided as a web server which is freely accessible at http://metagenomics.iiserb.ac.in/antiinflam/ .
目前针对炎症和自身免疫性疾病的治疗方法包括使用非特异性抗炎药物和其他免疫抑制剂,这些药物常常伴有潜在的副作用。作为一种替代疗法,抗炎肽最近正被开发用作治疗各种炎症性疾病(如阿尔茨海默病和类风湿性关节炎)的抗炎剂。因此,了解氨基酸序列与其潜在抗炎特性之间的相关性对于发现新型高效的基于抗炎肽的治疗方法至关重要。
在本研究中,我们开发了一种预测工具,用于将肽分类为抗炎表位或非抗炎表位。使用从免疫表位数据库和分析资源数据库获得的经过实验验证的表位进行训练。采用不同的基于序列的特征及其与基序信息的混合特征来开发基于支持向量机的机器学习模型。同样,也使用随机森林构建了机器学习模型。
肽的组成和末端残基保守性分析表明,与非抗炎表位相比,抗炎表位中亮氨酸、丝氨酸、酪氨酸和精氨酸残基占主导地位。同样,发现抗炎表位特异性基序富含疏水和极性残基。基于三肽组成的支持向量机模型与基序的混合在10倍交叉验证中表现最佳,准确率为78.1%,马修斯相关系数为0.58。在验证数据集上,其准确率为72%,马修斯相关系数为0.45,排除了过拟合的任何可能性。基于三肽组成的随机森林模型在10倍交叉验证中的准确率为0.8,马修斯相关系数为0.59,然而,与支持向量机模型相比,其在验证数据集上的准确率(0.68)和马修斯相关系数(0.31)较低。因此,支持向量机模型被用作默认模型,并提供了使用随机森林模型的附加选项。
预测模型以及表位映射和相似性搜索工具已作为一个网络服务器提供,可通过http://metagenomics.iiserb.ac.in/antiinflam/免费访问。