Koca Tuba Tulay
Department of Physical Medicine and Rehabilitation, Malatya State Hospital, Malatya, Turkey.
North Clin Istanb. 2016 May 14;3(2):135-139. doi: 10.14744/nci.2015.30602. eCollection 2016.
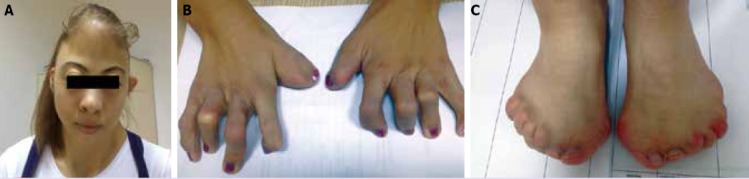
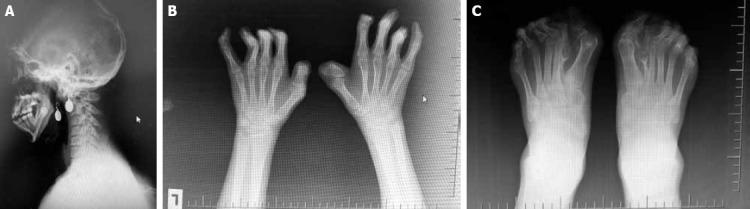
Apert syndrome is the rare acrocephalosyndactyly syndrome type 1, characterized by craniosynostosis, severe syndactyly of hands and feet, and dysmorphic facial features. It demonstrates autosomal dominant inheritance assigned to mutations in the fibroblast growth factor receptor gene. Presently described is case of a 19-year-old female patient diagnosed on physical examination with Apert syndrome based on acrocephaly, prominent forehead, ocular hypertelorism, proptosis, short and broad nose, pseudoprognathism, dental crowding and ectopia, maxillar hypoplasia, low hairline, webbed neck, pectus excavatum, and severe, bilateral syndactyly of hands and feet. The multiple phenotypic signs of Apert syndrome make multidisciplinary team, including dentist, neurosurgeon, plastic surgeon, physiatrist, ophthalmologist, perinatalogist and geneticist, essential for successful management.
Apert综合征是罕见的1型尖头并指综合征,其特征为颅缝早闭、手足严重并指以及面部畸形特征。它表现为常染色体显性遗传,由成纤维细胞生长因子受体基因突变所致。本文介绍了一名19岁女性患者的病例,该患者经体格检查诊断为Apert综合征,依据为尖头畸形、前额突出、眼距增宽、眼球突出、短而宽的鼻子、假性下颌前突、牙齿拥挤和异位、上颌骨发育不全、低发际线、蹼颈、漏斗胸以及严重的双侧手足并指。Apert综合征的多种表型体征使得多学科团队(包括牙医、神经外科医生、整形外科医生、物理治疗师、眼科医生、围产医学专家和遗传学家)对于成功治疗至关重要。