Division of Immunogenetics, Department of Immunobiology and Neuroscience, Medical Institute of Bioregulation, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582, Japan.
Department of Dermatology, Graduate School of Medical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582, Japan.
Nat Commun. 2017 Jan 9;8:13946. doi: 10.1038/ncomms13946.
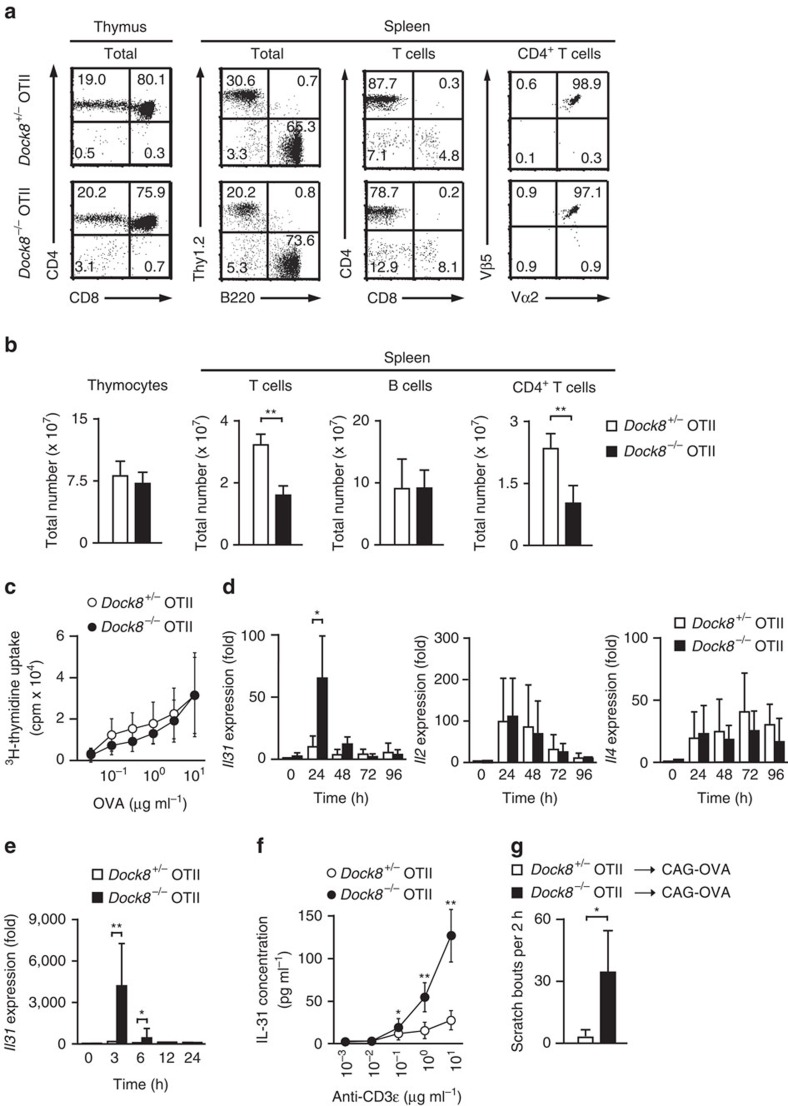
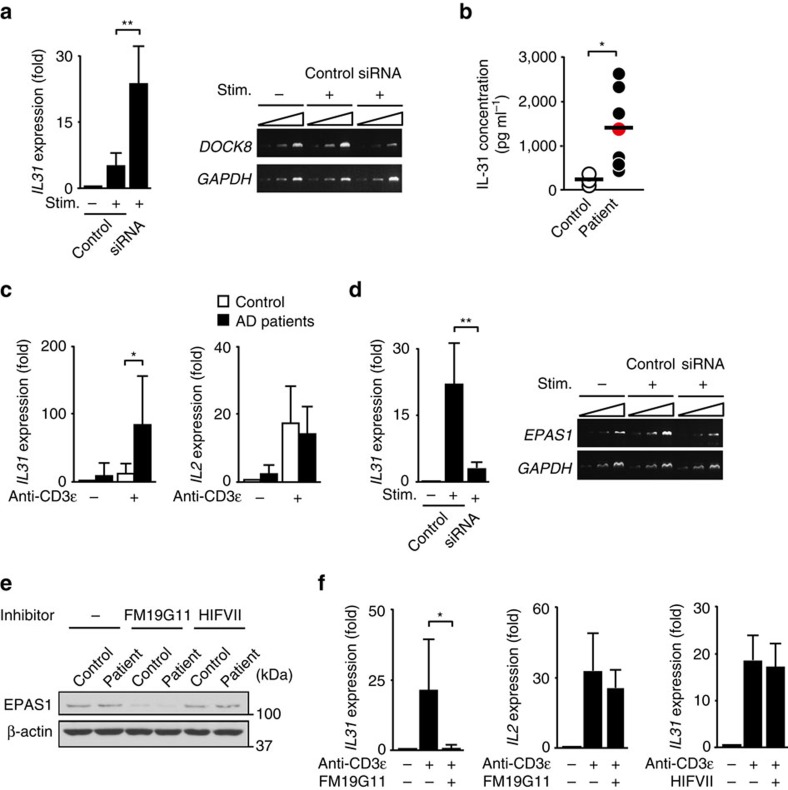
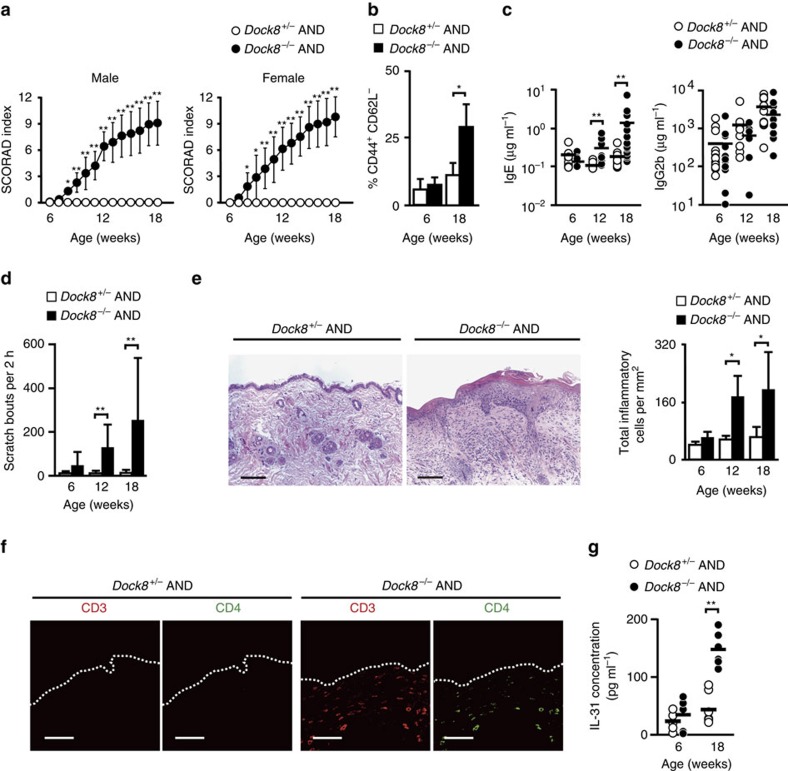
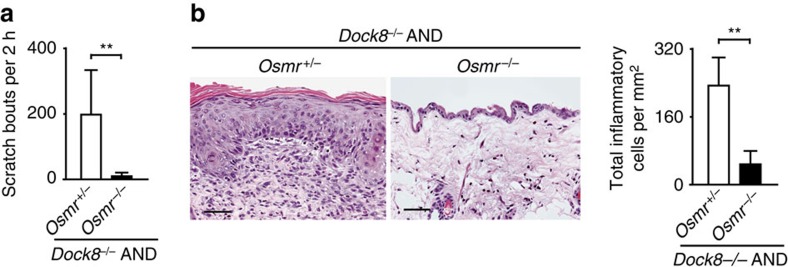
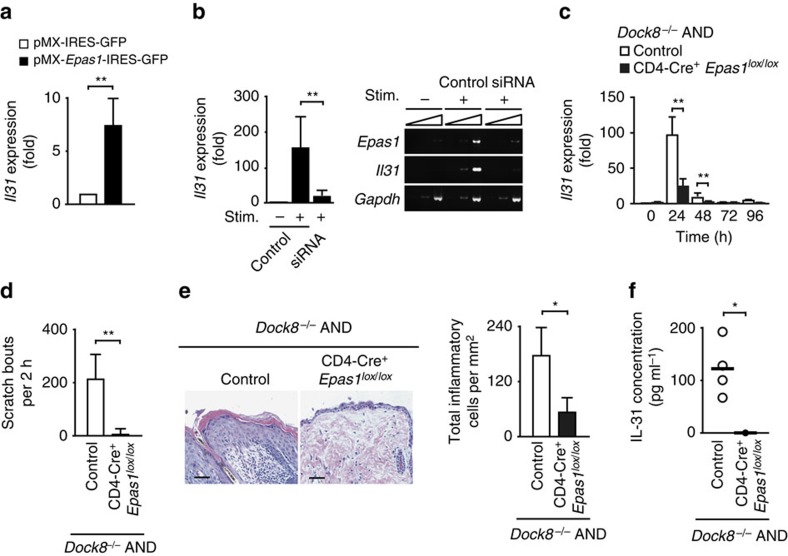
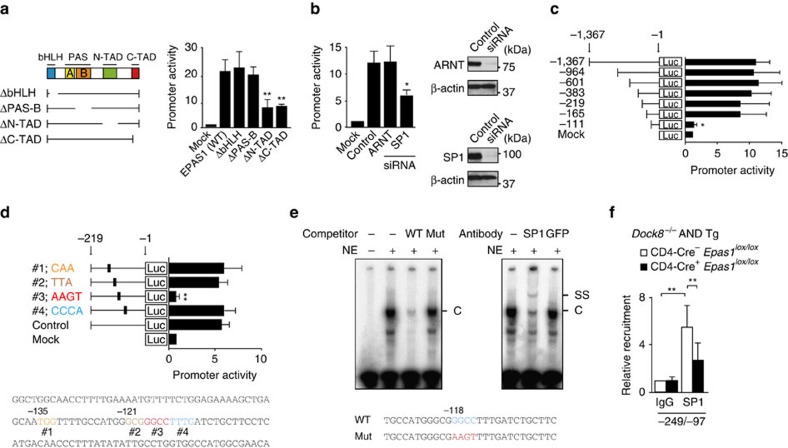
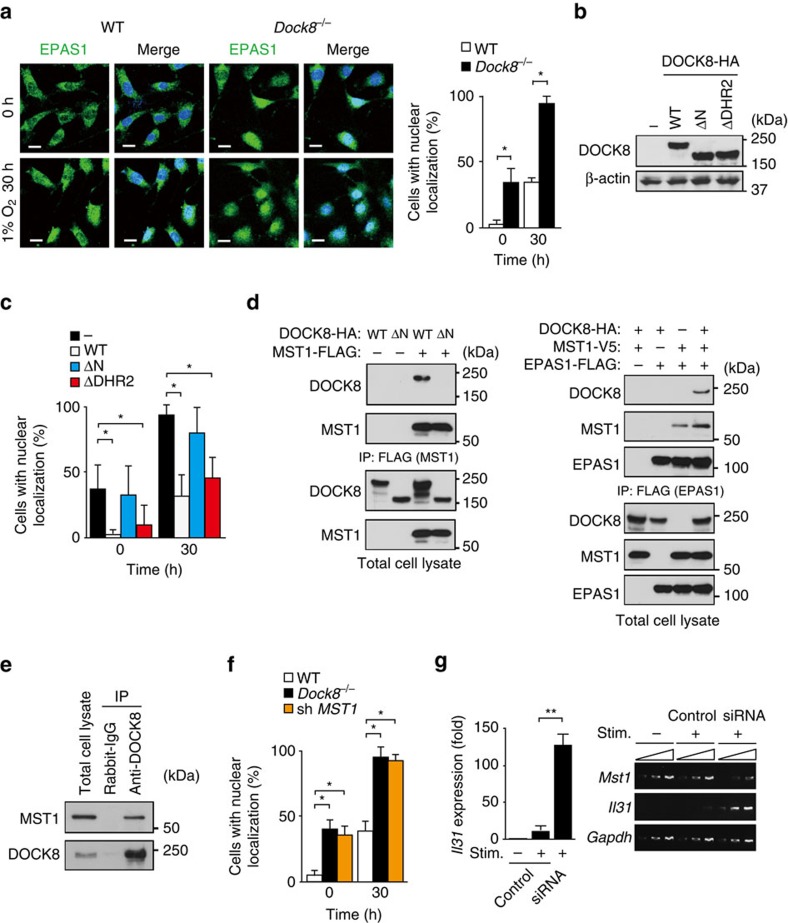
Mutations of DOCK8 in humans cause a combined immunodeficiency characterized by atopic dermatitis with high serum IgE levels. However, the molecular link between DOCK8 deficiency and atopic skin inflammation is unknown. Here we show that CD4 T cells from DOCK8-deficient mice produce large amounts of IL-31, a major pruritogen associated with atopic dermatitis. IL-31 induction critically depends on the transcription factor EPAS1, and its conditional deletion in CD4 T cells abrogates skin disease development in DOCK8-deficient mice. Although EPAS1 is known to form a complex with aryl hydrocarbon receptor nuclear translocator (ARNT) and control hypoxic responses, EPAS1-mediated Il31 promoter activation is independent of ARNT, but in collaboration with SP1. On the other hand, we find that DOCK8 is an adaptor and negative regulator of nuclear translocation of EPAS1. Thus, EPAS1 links DOCK8 deficiency to atopic skin inflammation via IL-31 induction in CD4 T cells.
人类 DOCK8 突变导致以特应性皮炎和高血清 IgE 水平为特征的联合免疫缺陷。然而,DOCK8 缺乏和特应性皮肤炎症之间的分子联系尚不清楚。在这里,我们显示 DOCK8 缺陷小鼠的 CD4 T 细胞产生大量与特应性皮炎相关的主要瘙痒原 IL-31。IL-31 的诱导严重依赖于转录因子 EPAS1,其在 CD4 T 细胞中的条件缺失可消除 DOCK8 缺陷小鼠的皮肤疾病发展。尽管众所周知 EPAS1 与芳香烃受体核转位蛋白 (ARNT) 形成复合物并控制低氧反应,但 EPAS1 介导的 Il31 启动子激活不依赖于 ARNT,而是与 SP1 协作。另一方面,我们发现 DOCK8 是 EPAS1 核易位的衔接蛋白和负调节剂。因此,EPAS1 通过 CD4 T 细胞中 IL-31 的诱导将 DOCK8 缺陷与特应性皮肤炎症联系起来。