Shen Xiao, Nguyen Thach T, Koh Ming Joo, Xu Dongmin, Speed Alexander W H, Schrock Richard R, Hoveyda Amir H
Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, USA.
Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA.
Nature. 2017 Jan 19;541(7637):380-385. doi: 10.1038/nature20800. Epub 2017 Jan 9.
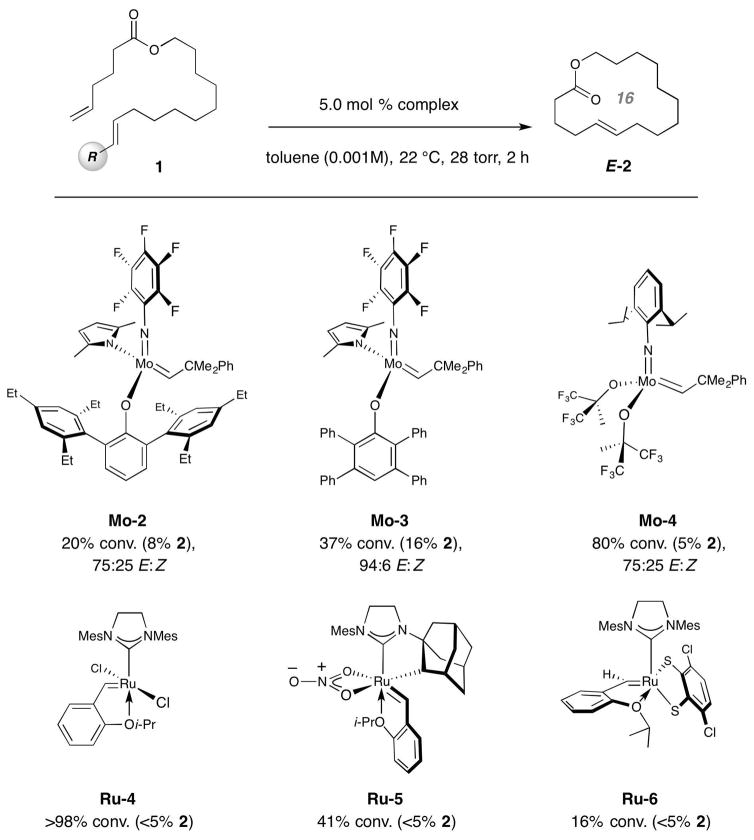
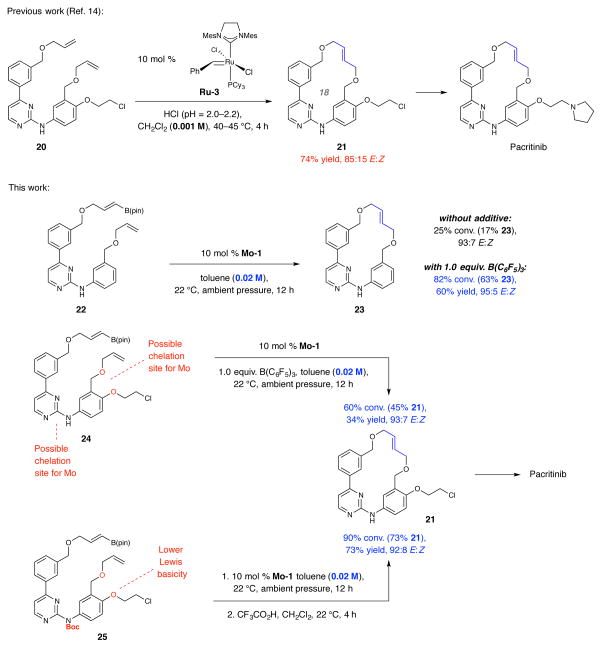
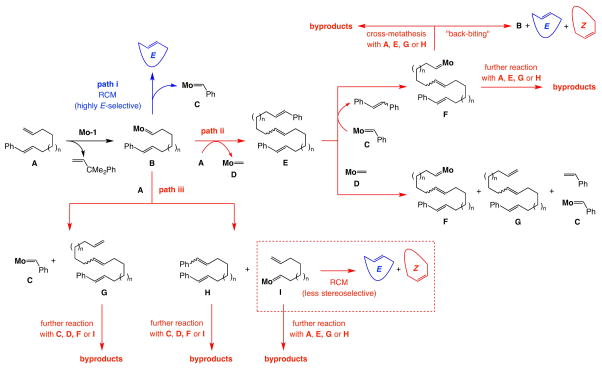
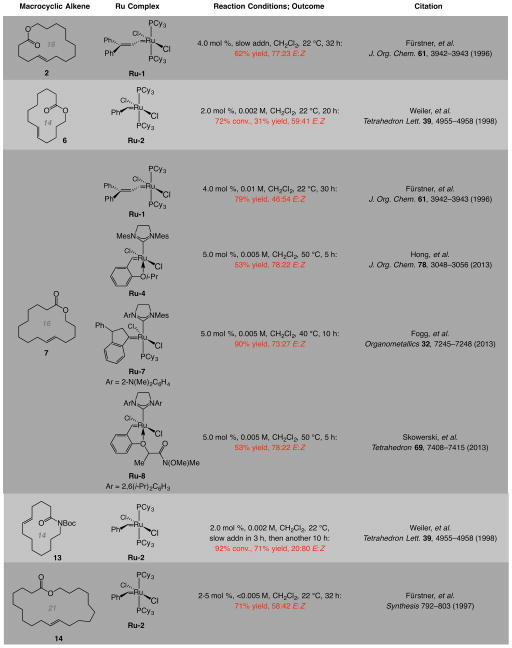
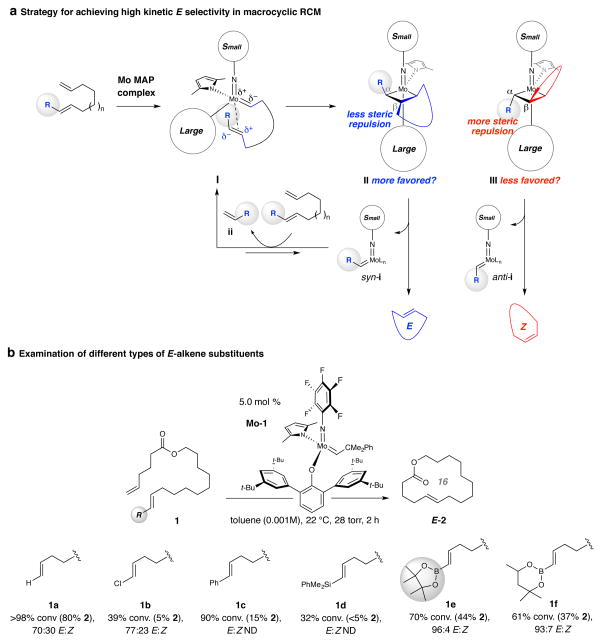
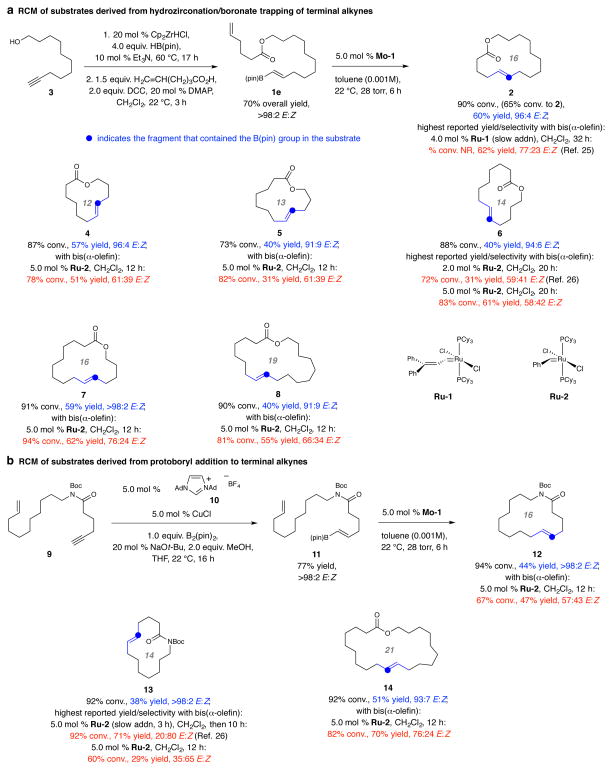
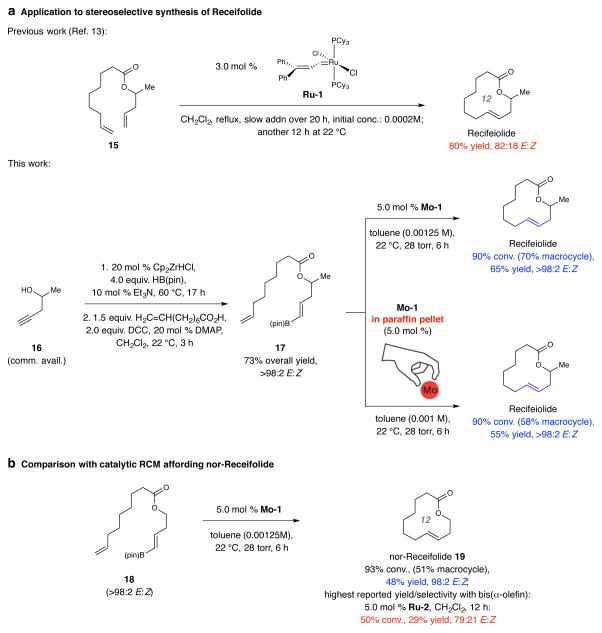
Macrocyclic compounds are central to the development of new drugs, but preparing them can be challenging because of the energy barrier that must be surmounted in order to bring together and fuse the two ends of an acyclic precursor such as an alkene (also known as an olefin). To this end, the catalytic process known as ring-closing metathesis (RCM) has allowed access to countless biologically active macrocyclic organic molecules, even for large-scale production. Stereoselectivity is often critical in such cases: the potency of a macrocyclic compound can depend on the stereochemistry of its alkene; alternatively, one isomer of the compound can be subjected to stereoselective modification (such as dihydroxylation). Kinetically controlled Z-selective RCM reactions have been reported, but the only available metathesis approach for accessing macrocyclic E-olefins entails selective removal of the Z-component of a stereoisomeric mixture by ethenolysis, sacrificing substantial quantities of material if E/Z ratios are near unity. Use of ethylene can also cause adventitious olefin isomerization-a particularly serious problem when the E-alkene is energetically less favoured. Here, we show that dienes containing an E-alkenyl-B(pinacolato) group, widely used in catalytic cross-coupling, possess the requisite electronic and steric attributes to allow them to be converted stereoselectively to E-macrocyclic alkenes. The reaction is promoted by a molybdenum monoaryloxide pyrrolide complex and affords products at a yield of up to 73 per cent and an E/Z ratio greater than 98/2. We highlight the utility of the approach by preparing recifeiolide (a 12-membered-ring antibiotic) and pacritinib (an 18-membered-ring enzyme inhibitor), the Z-isomer of which is less potent than the E-isomer. Notably, the 18-membered-ring moiety of pacritinib-a potent anti-cancer agent that is in advanced clinical trials for treating lymphoma and myelofibrosis-was prepared by RCM carried out at a substrate concentration 20 times greater than when a ruthenium carbene was used.
大环化合物是新药研发的核心,但由于在将无环前体(如烯烃,也称为烯属烃)的两端聚集并融合时必须克服能量障碍,制备它们可能具有挑战性。为此,称为闭环复分解(RCM)的催化过程使得能够获得无数具有生物活性的大环有机分子,甚至可用于大规模生产。在这种情况下,立体选择性通常至关重要:大环化合物的效力可能取决于其烯烃的立体化学;或者,化合物的一种异构体可进行立体选择性修饰(如二羟基化)。已经报道了动力学控制的Z选择性RCM反应,但用于获得大环E-烯烃的唯一可用复分解方法是通过乙烯解选择性去除立体异构混合物的Z组分,如果E/Z比接近1,则会损失大量材料。使用乙烯还可能导致意外的烯烃异构化——当E-烯烃在能量上不太有利时,这是一个特别严重的问题。在这里,我们表明,在催化交叉偶联中广泛使用的含有E-烯基-B(频哪醇硼酸酯)基团的二烯具有必要的电子和空间属性,使其能够立体选择性地转化为E-大环烯烃。该反应由单芳氧基钼吡咯配合物促进,产率高达73%,E/Z比大于98/2。我们通过制备瑞西奥内酯(一种12元环抗生素)和帕西替尼(一种18元环酶抑制剂)突出了该方法的实用性,其Z异构体的效力低于E异构体。值得注意的是,帕西替尼的18元环部分——一种在治疗淋巴瘤和骨髓纤维化的晚期临床试验中的强效抗癌剂——是通过RCM制备的,其底物浓度是使用卡宾钌时的20倍。