Chiong Mary Anne D, Canson Daffodil M, Abacan Mary Ann R, Baluyot Melissa Mae P, Cordero Cynthia P, Silao Catherine Lynn T
Institute of Human Genetics, National Institutes of Health, University of the Philippines Manila, 625 Pedro Gil St., Ermita, Manila, 1000, Philippines.
Department of Pediatrics, University of the Philippines-Philippine General Hospital, Manila, Philippines.
Orphanet J Rare Dis. 2017 Jan 11;12(1):7. doi: 10.1186/s13023-016-0558-0.
Mucopolysaccharidosis type II, an X-linked recessive disorder is the most common lysosomal storage disease detected among Filipinos. This is a case series involving 23 male Filipino patients confirmed to have Hunter syndrome. The clinical and biochemical characteristics were obtained and mutation testing of the IDS gene was done on the probands and their female relatives.
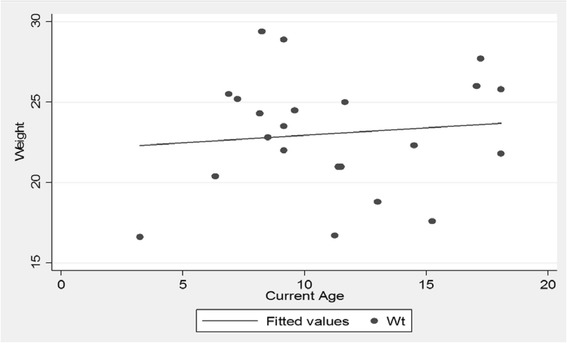
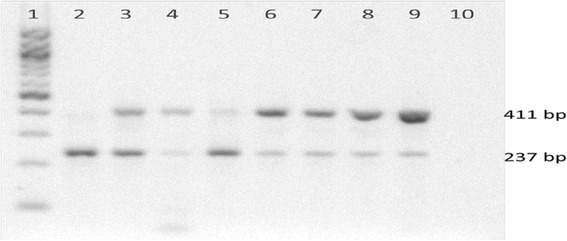
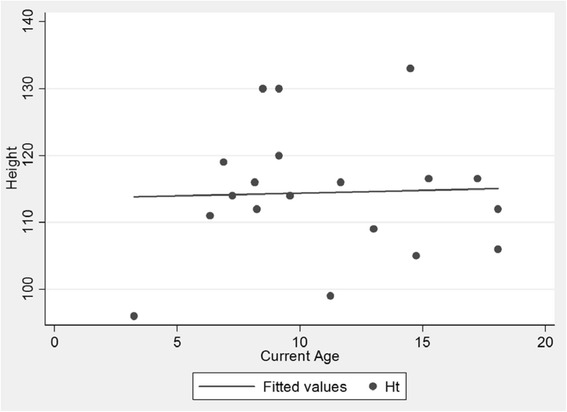
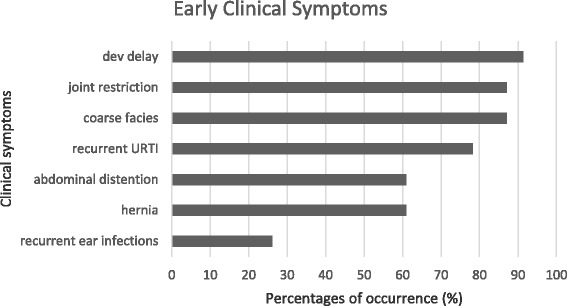
The mean age of the patients was 11.28 (SD 4.10) years with an average symptom onset at 1.2 (SD 1.4) years. The mean age at biochemical diagnosis was 8 (SD 3.2) years. The early clinical characteristics were developmental delay, joint stiffness, coarse facies, recurrent respiratory tract infections, abdominal distention and hernia. Majority of the patients had joint contractures, severe intellectual disability, error of refraction, hearing loss and valvular regurgitation on subspecialists' evaluation. The mean GAG concentration was 506.5 mg (SD 191.3)/grams creatinine while the mean plasma iduronate-2-sulfatase activity was 0.86 (SD 0.79) nmol/mg plasma/4 h. Fourteen (14) mutations were found: 6 missense (42.9%), 4 nonsense (28.6%), 2 frameshift (14.3%), 1 exon skipping at the cDNA level (7.1%), and 1 gross insertion (7.1%). Six (6) novel mutations were observed (43%): p.C422F, p.P86Rfs44, p.Q121, p.L209Wfs*4, p.T409R, and c.1461_1462insN[710].
The age at diagnosis in this series was much delayed and majority of the patients presented with severe neurologic impairment. The results of the biochemical tests did not contribute to the phenotypic classification of patients. The effects of the mutations were consistent with the severe phenotype seen in the majority of the patients.
II型粘多糖贮积症是一种X连锁隐性疾病,是菲律宾人中最常见的溶酶体贮积病。这是一个病例系列,涉及23名确诊患有亨特综合征的菲律宾男性患者。获取了临床和生化特征,并对先证者及其女性亲属进行了IDS基因的突变检测。
患者的平均年龄为11.28(标准差4.10)岁,平均症状出现年龄为1.2(标准差1.4)岁。生化诊断的平均年龄为8(标准差3.2)岁。早期临床特征为发育迟缓、关节僵硬、面容粗糙、反复呼吸道感染、腹胀和疝气。在专科评估中,大多数患者有关节挛缩、严重智力残疾、屈光不正、听力丧失和瓣膜反流。平均糖胺聚糖浓度为506.5毫克(标准差191.3)/克肌酐,而平均血浆艾杜糖醛酸-2-硫酸酯酶活性为0.86(标准差0.79)纳摩尔/毫克血浆/4小时。发现了14种突变:6种错义突变(42.9%)、4种无义突变(28.6%)、2种移码突变(14.3%)、1种cDNA水平的外显子跳跃(7.1%)和1种大片段插入(7.1%)。观察到6种(6)新突变(43%):p.C422F、p.P86Rfs44、p.Q121、p.L209Wfs*4、p.T409R和c.1461_1462insN[710]。
该系列中的诊断年龄延迟很多,大多数患者表现出严重的神经功能损害。生化检测结果对患者的表型分类没有帮助。突变的影响与大多数患者中观察到的严重表型一致。