Puttick Mark N, O'Reilly Joseph E, Tanner Alastair R, Fleming James F, Clark James, Holloway Lucy, Lozano-Fernandez Jesus, Parry Luke A, Tarver James E, Pisani Davide, Donoghue Philip C J
School of Earth Sciences, University of Bristol, Life Sciences Building, 24 Tyndall Avenue, Bristol BS8 1TQ, UK.
Department of Life Sciences, Natural History Museum, Cromwell Road, London SW7 5BD, UK.
Proc Biol Sci. 2017 Jan 11;284(1846). doi: 10.1098/rspb.2016.2290.
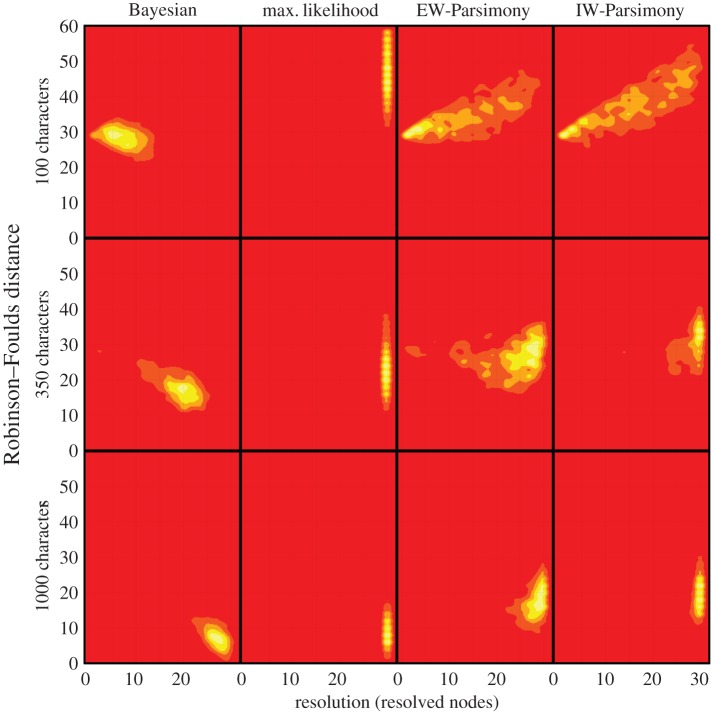
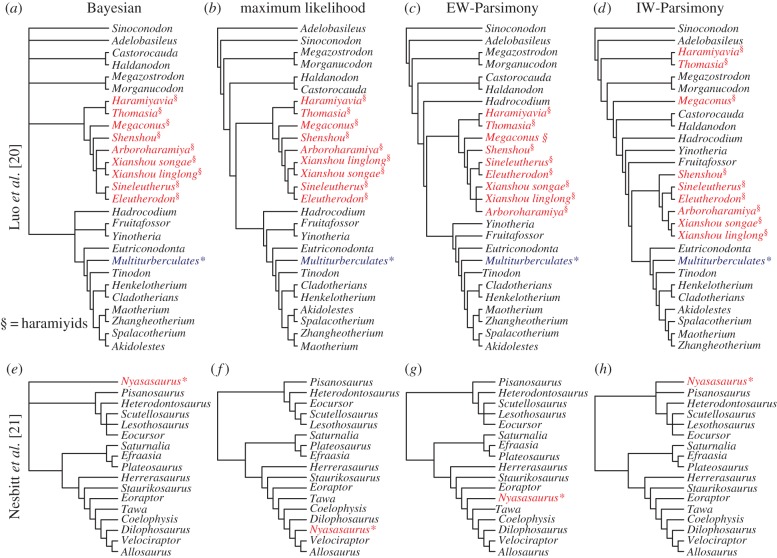
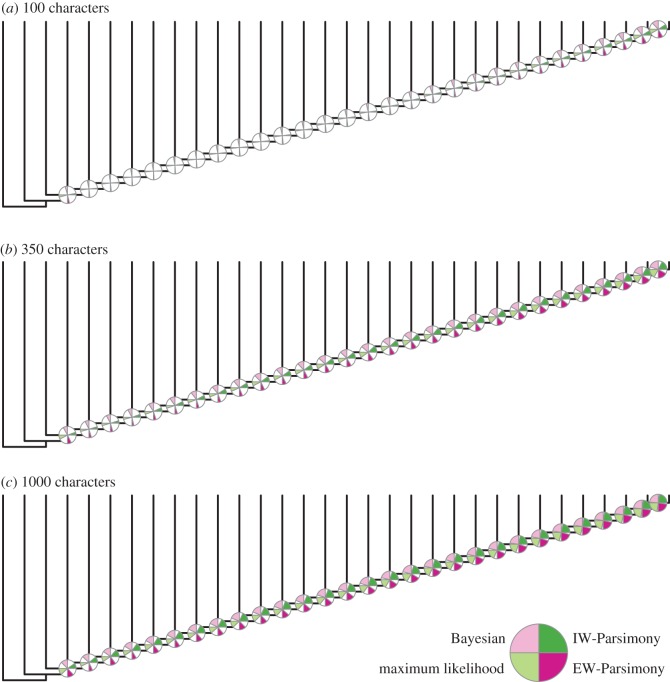
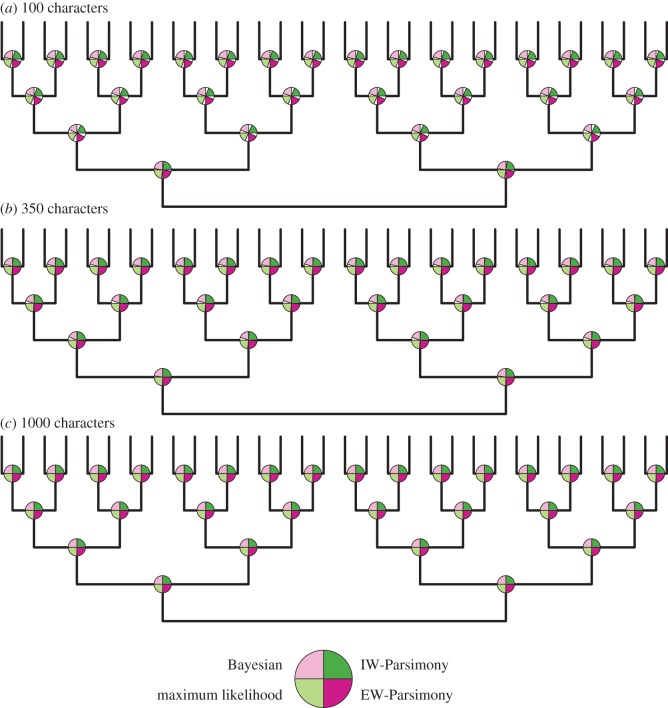
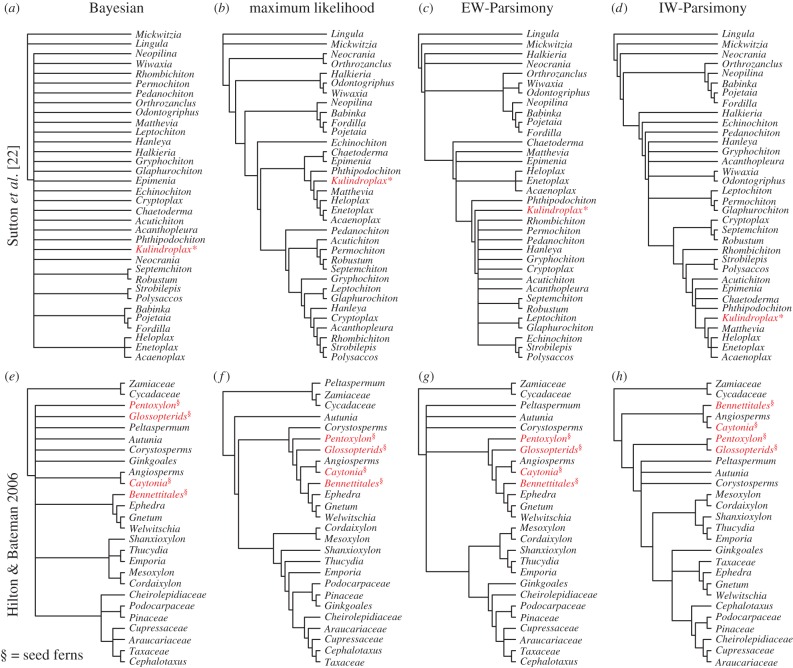
Morphological data provide the only means of classifying the majority of life's history, but the choice between competing phylogenetic methods for the analysis of morphology is unclear. Traditionally, parsimony methods have been favoured but recent studies have shown that these approaches are less accurate than the Bayesian implementation of the Mk model. Here we expand on these findings in several ways: we assess the impact of tree shape and maximum-likelihood estimation using the Mk model, as well as analysing data composed of both binary and multistate characters. We find that all methods struggle to correctly resolve deep clades within asymmetric trees, and when analysing small character matrices. The Bayesian Mk model is the most accurate method for estimating topology, but with lower resolution than other methods. Equal weights parsimony is more accurate than implied weights parsimony, and maximum-likelihood estimation using the Mk model is the least accurate method. We conclude that the Bayesian implementation of the Mk model should be the default method for phylogenetic estimation from phenotype datasets, and we explore the implications of our simulations in reanalysing several empirical morphological character matrices. A consequence of our finding is that high levels of resolution or the ability to classify species or groups with much confidence should not be expected when using small datasets. It is now necessary to depart from the traditional parsimony paradigms of constructing character matrices, towards datasets constructed explicitly for Bayesian methods.
形态学数据是划分大多数生命历史的唯一手段,但在用于分析形态学的相互竞争的系统发育方法之间做出选择并不明确。传统上,简约方法备受青睐,但最近的研究表明,这些方法不如Mk模型的贝叶斯实现准确。在这里,我们从几个方面扩展了这些发现:我们评估树形和使用Mk模型的最大似然估计的影响,以及分析由二元和多状态特征组成的数据。我们发现,在分析不对称树中的深支以及分析小特征矩阵时,所有方法都难以正确解析。贝叶斯Mk模型是估计拓扑结构最准确的方法,但分辨率低于其他方法。等权重简约法比隐含权重简约法更准确,使用Mk模型的最大似然估计是最不准确的方法。我们得出结论,Mk模型的贝叶斯实现应该是从表型数据集中进行系统发育估计的默认方法,并且我们探讨了我们的模拟在重新分析几个经验形态特征矩阵中的意义。我们发现的一个结果是,在使用小数据集时,不应期望有高水平的分辨率或有很大把握对物种或类群进行分类的能力。现在有必要背离构建特征矩阵的传统简约范式,转向为贝叶斯方法明确构建的数据集。