Zhou Ning, Ye Yong, Wang Xingxu, Ma Ben, Wu Jian, Li Lei, Wang Lin, Wang Dao Wen, Zou Yunzeng
Division of Cardiology, Department of Internal Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Avenue, Wuhan, 430030, China.
Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital and Institutes of Biological Science, Fudan University, Shanghai, 200032, China.
J Mol Med (Berl). 2017 Apr;95(4):445-460. doi: 10.1007/s00109-016-1504-2. Epub 2017 Jan 13.
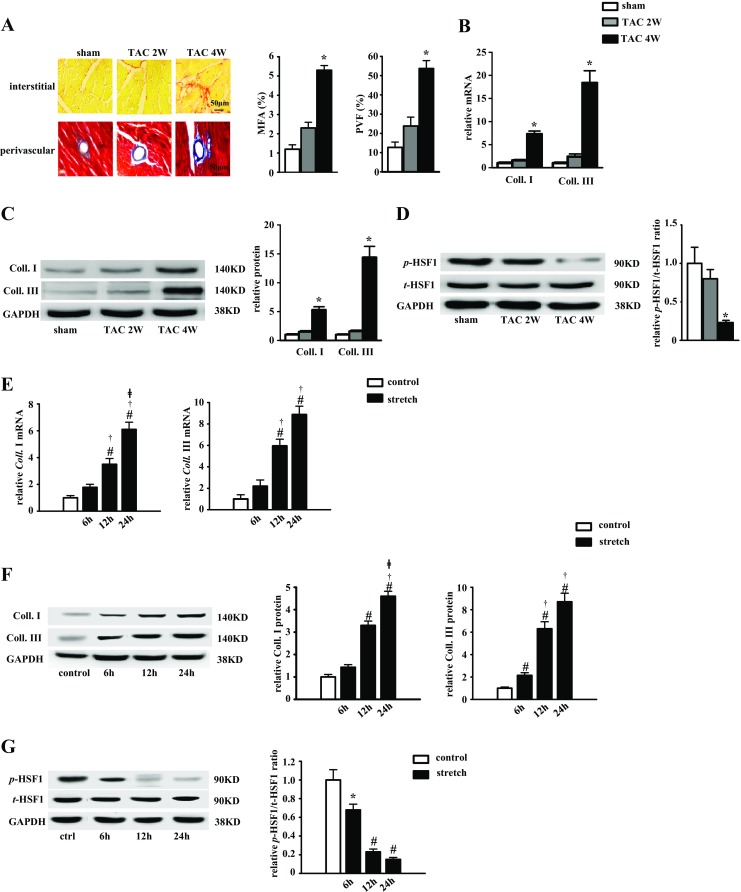
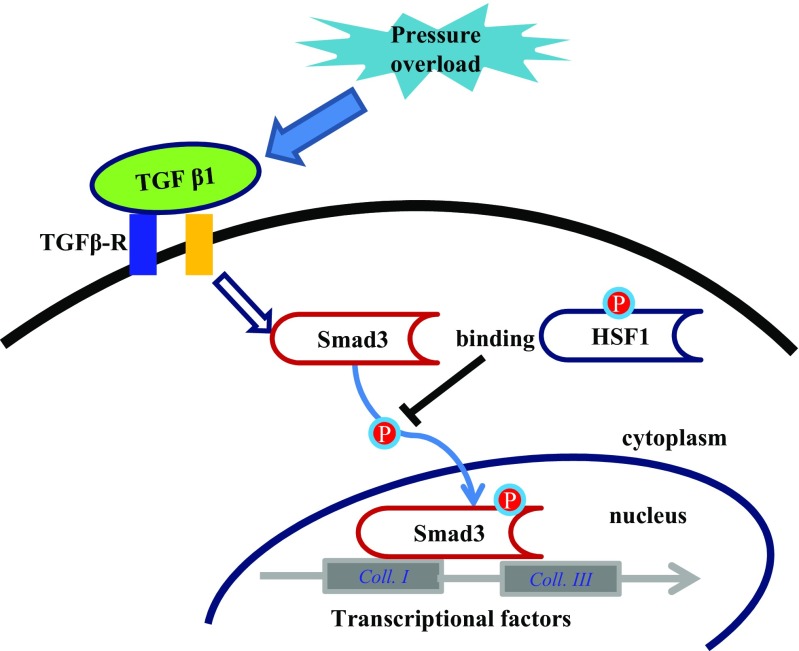
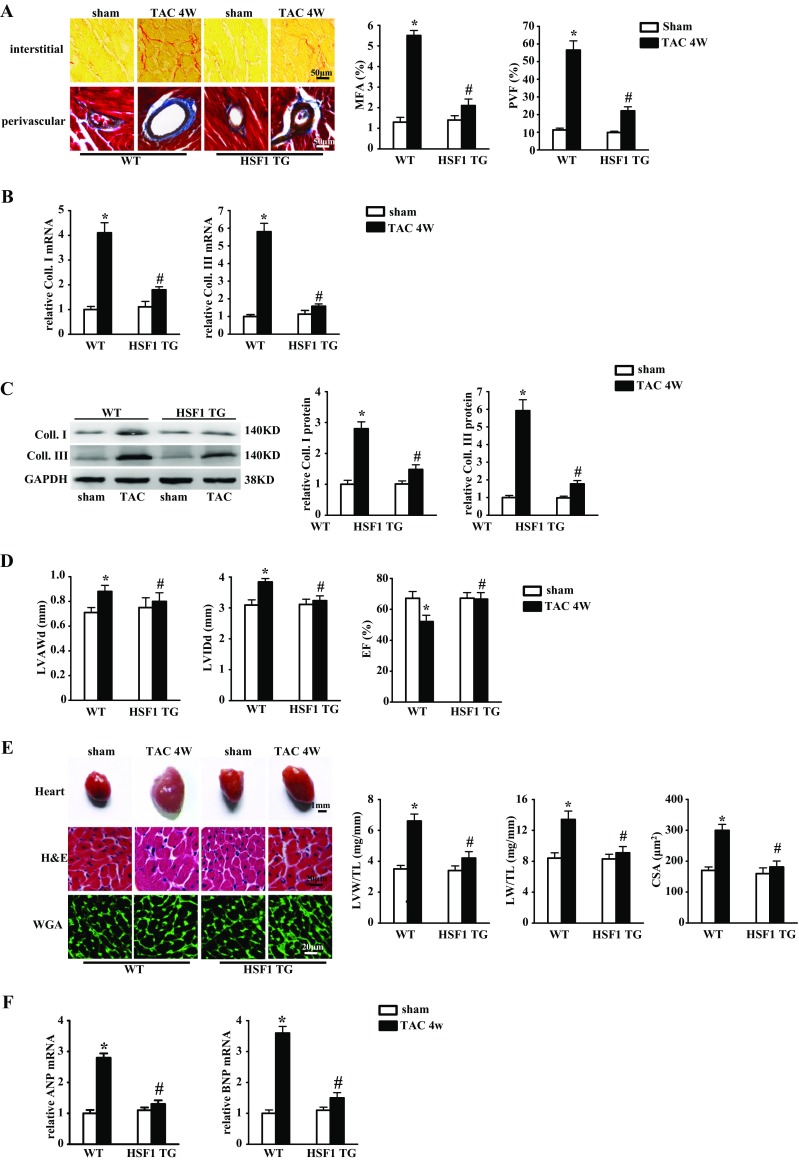
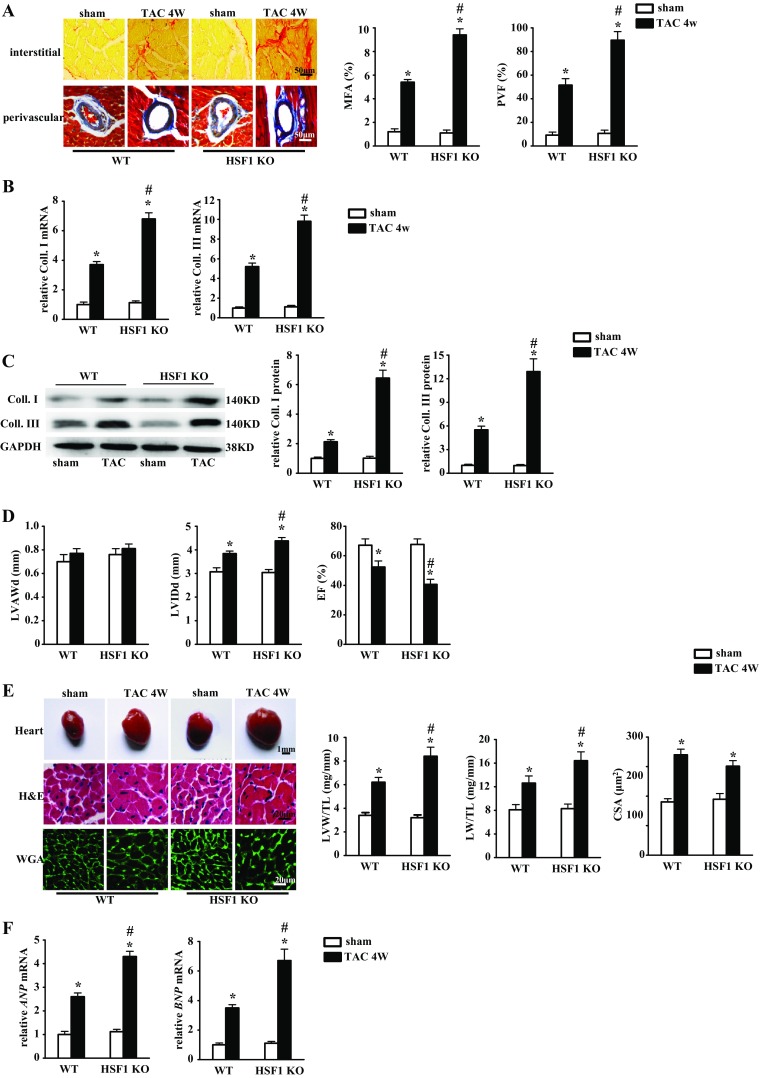
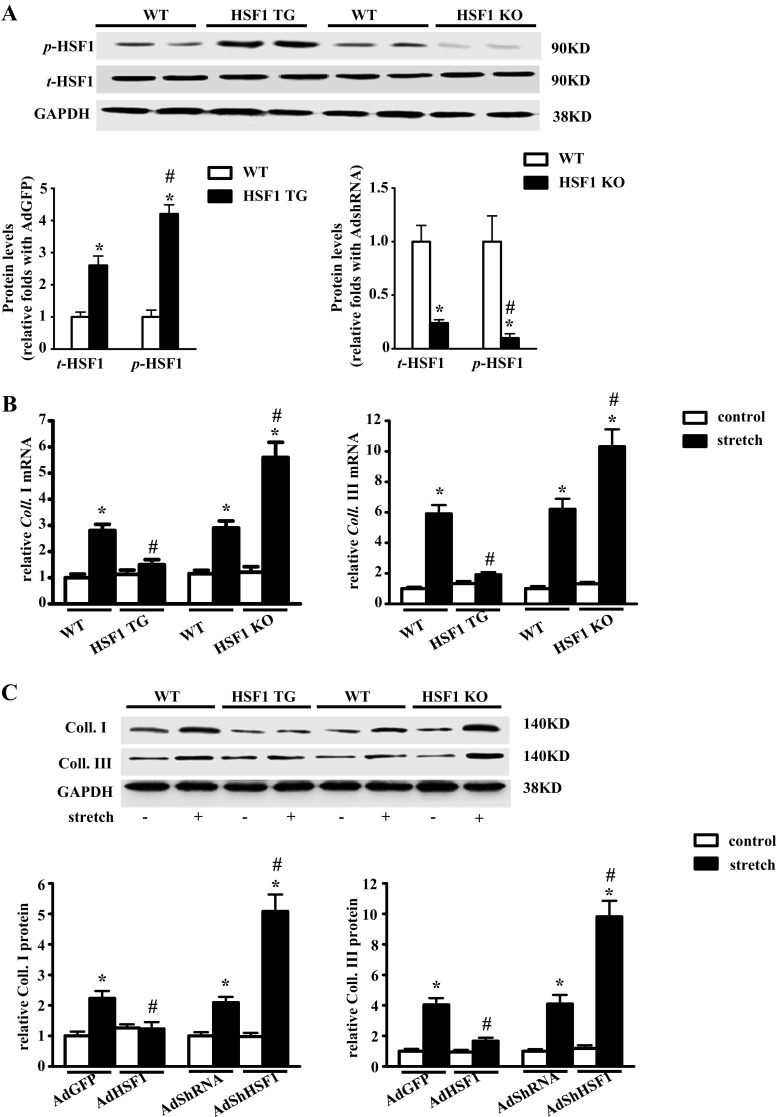
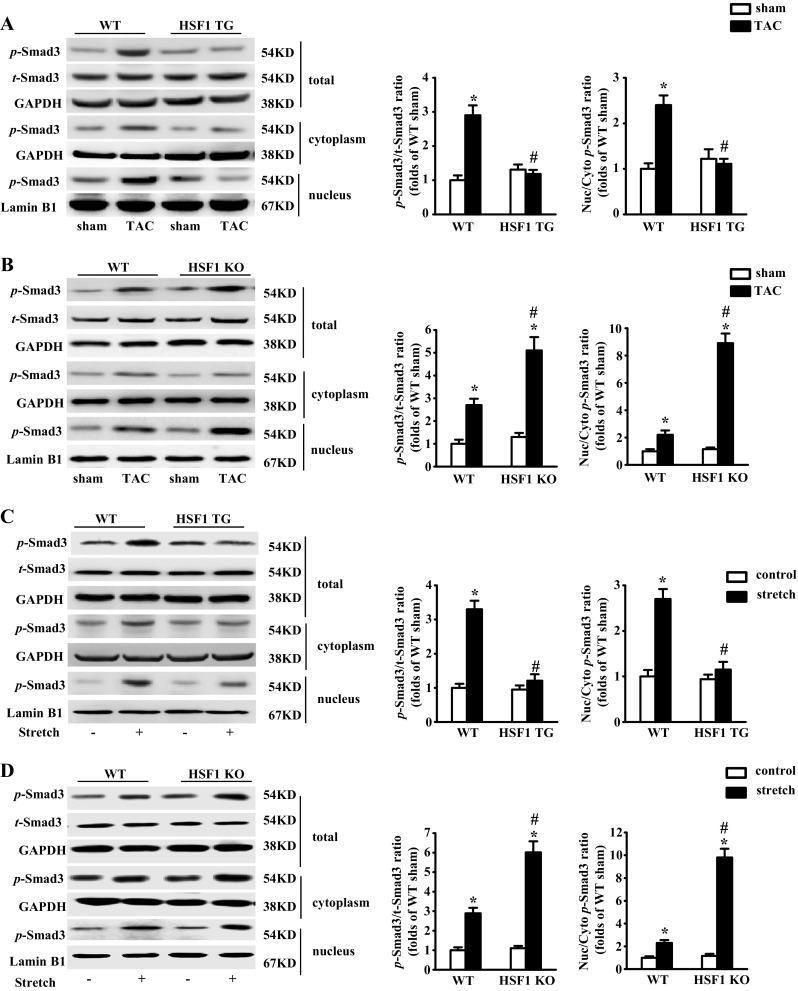
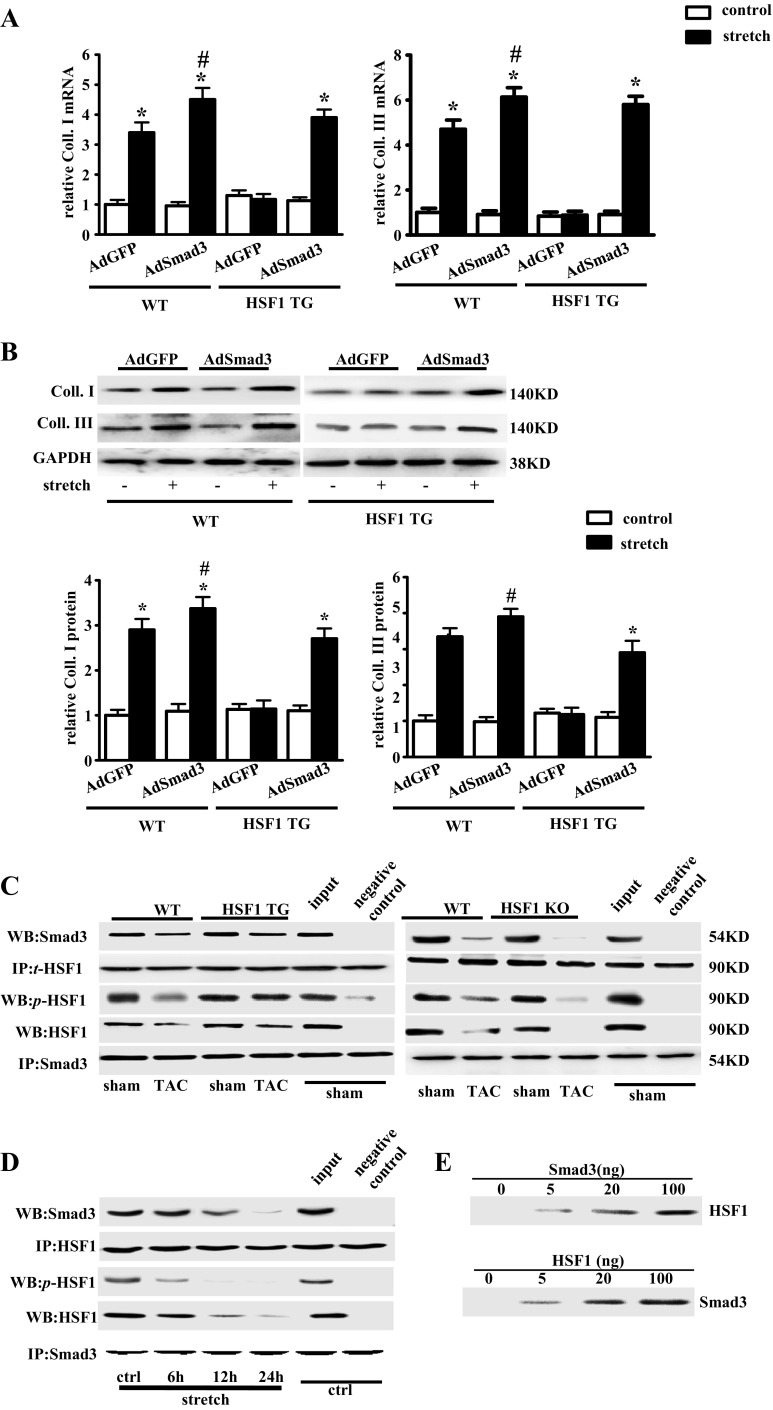
Fibrotic cardiac muscle exhibits high stiffness and low compliance which are major risk factors of heart failure. Although heat shock transcription factor 1 (HSF1) was identified as an intrinsic cardioprotective factor, the role that HSF1 plays in cardiac fibrosis remains unclear. Our study aims to investigate the role of HSF1 in pressure overload-induced cardiac fibrosis and the underlying mechanism. HSF1 phosphorylation was significantly downregulated in transverse aortic constriction (TAC)-treated mouse hearts and mechanically stretched cardiac fibroblasts (cFBs). HSF1 transgenic (TG) mice, HSF1 deficient heterozygote (KO) mice, and their wild-type littermates were subjected to sham or TAC surgery for 4 weeks. HSF1 overexpression significantly attenuated pressure overload-induced cardiac fibrosis and dysfunction. Conversely, HSF1 KO mice showed deteriorated fibrotic response and cardiac dysfunction upon TAC. Moreover, we uncovered that overexpression of HSF1 protected against fibrotic response of cFBs to pressure overload. Mechanistically, we observed that the phosphorylation and the nuclear distribution of the Smad family member 3 (Smad3) were significantly decreased in HSF1-overexpressing mouse hearts, while being greatly increased in HSF1 KO mouse hearts upon TAC, compared to the control hearts, respectively. Similar alteration of Smad3 phosphorylation and nuclear distribution were found in isolated mouse cardiac fibroblasts and mechanically stretched cFBs. Constitutively active Smad3 blocked the anti-fibrotic effect of HSF1 in cFBs. Furthermore, we found a direct binding of phosphorylated HSF1 and Smad3, which can be suppressed by mechanical stress. In conclusion, the present study demonstrated for the first time that HSF1 acts as a novel negative regulator of cardiac fibrosis by blocking Smad3 activation.
HSF1 activity is decreased in fibrotic hearts. HSF1 overexpression attenuates pressure overload-induced cardiac fibrosis and dysfunction. Deficiency of HSF1 deteriorates fibrotic response and cardiac dysfunction upon TAC. HSF1 inhibits phosphorylation and nuclear distribution of Smad3 via direct binding to Smad3. Active Smad3 blocks the anti-fibrotic effect of HSF1.
纤维化心肌表现出高僵硬度和低顺应性,这是心力衰竭的主要危险因素。尽管热休克转录因子1(HSF1)被确定为一种内在的心脏保护因子,但其在心脏纤维化中的作用仍不清楚。我们的研究旨在探讨HSF1在压力超负荷诱导的心脏纤维化中的作用及其潜在机制。在经主动脉缩窄(TAC)处理的小鼠心脏和机械拉伸的心脏成纤维细胞(cFBs)中,HSF1磷酸化显著下调。将HSF1转基因(TG)小鼠、HSF1缺陷杂合子(KO)小鼠及其野生型同窝小鼠进行假手术或TAC手术4周。HSF1过表达显著减轻压力超负荷诱导的心脏纤维化和功能障碍。相反,HSF1 KO小鼠在TAC后纤维化反应和心脏功能障碍恶化。此外,我们发现HSF1过表达可保护cFBs免受压力超负荷的纤维化反应。机制上,我们观察到与对照心脏相比,在HSF1过表达的小鼠心脏中,Smad家族成员3(Smad3)的磷酸化和核分布显著降低,而在TAC后的HSF1 KO小鼠心脏中则显著增加。在分离的小鼠心脏成纤维细胞和机械拉伸的cFBs中发现了Smad3磷酸化和核分布的类似改变。组成型活性Smad3阻断了HSF1在cFBs中的抗纤维化作用。此外,我们发现磷酸化的HSF1与Smad3直接结合,这可被机械应力抑制。总之,本研究首次证明HSF1通过阻断Smad3激活而作为心脏纤维化的新型负调节因子。
在纤维化心脏中HSF1活性降低。HSF1过表达减轻压力超负荷诱导的心脏纤维化和功能障碍。HSF1缺陷会使TAC后的纤维化反应和心脏功能障碍恶化。HSF1通过直接结合Smad3抑制Smad3的磷酸化和核分布。活性Smad3阻断HSF1的抗纤维化作用。