Lee Jae Young, Seo Dongyeob, You Jiyeon, Chung Sehee, Park Jin Seok, Lee Ji-Hyung, Jung Su Myung, Lee Youn Sook, Park Seok Hee
Department of Biological Sciences, Sungkyunkwan University, Suwon, Korea.
FEBS Lett. 2017 Feb;591(3):479-490. doi: 10.1002/1873-3468.12558. Epub 2017 Jan 29.
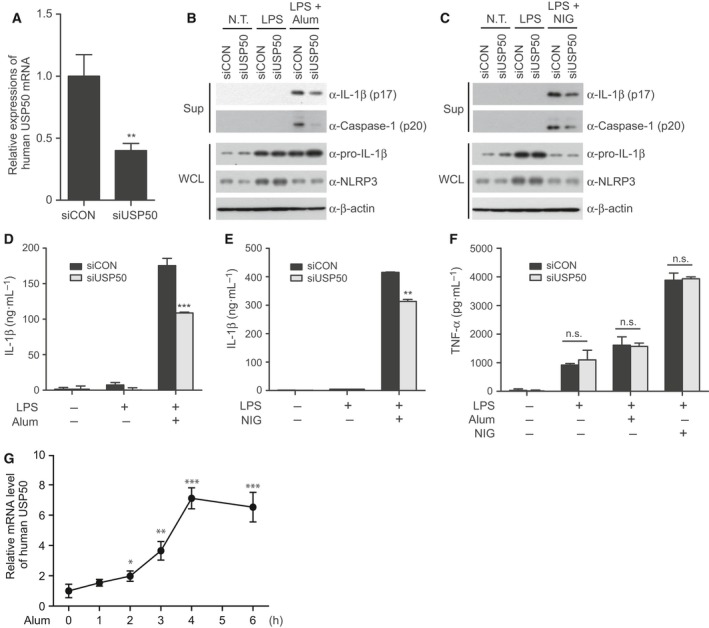
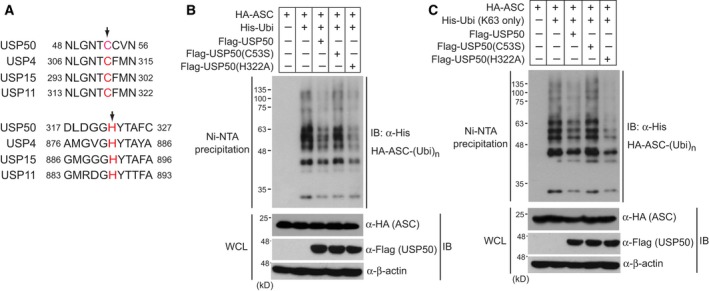
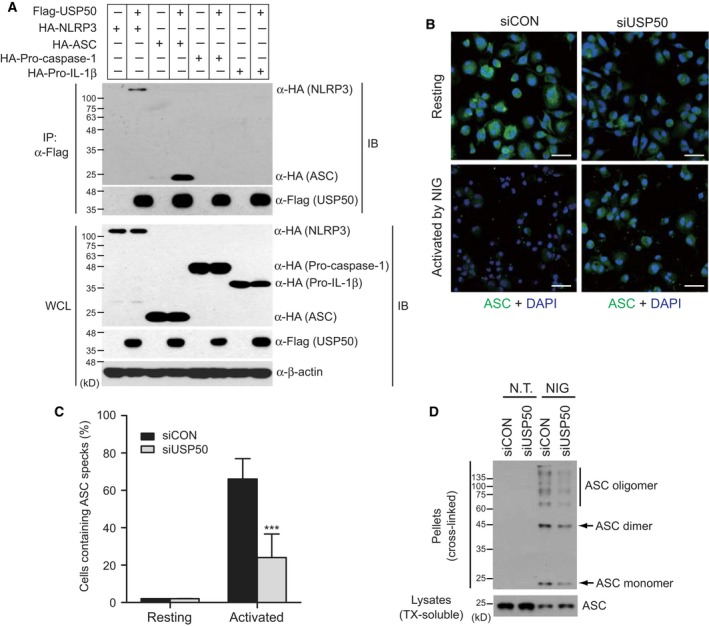
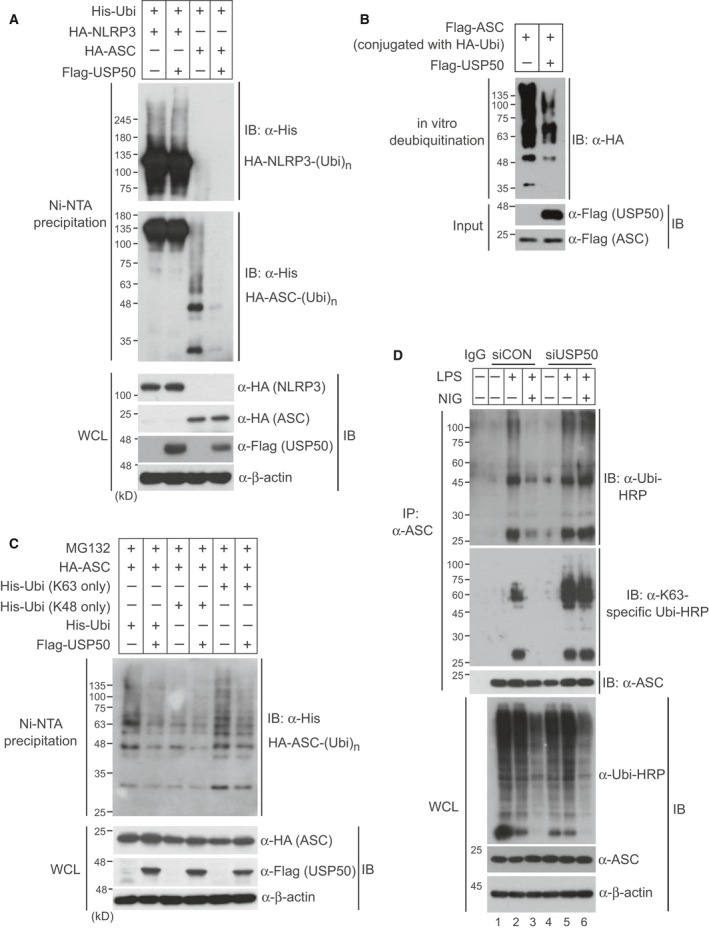
NOD-like receptor family protein 3 (NLRP3)-mediated inflammasome activation promotes caspase-1-dependent production of interleukin-1β (IL-1β) and requires the adaptor protein ASC. Compared with the priming and activation mechanisms of the inflammasome signaling pathway, post-translational ubiquitination/deubiquitination mechanisms controlling inflammasome activation have not been clearly addressed. We here demonstrate that the deubiquitinating enzyme USP50 binds to the ASC protein and subsequently regulates the inflammasome signaling pathway by deubiquitinating the lysine 63-linked polyubiquitination of ASC. USP50 knockdown in human THP-1 cells and mouse bone marrow-derived macrophages shows a significant decrease in procaspase-1 cleavage, resulting in a reduced secretion of IL-1β and interleukin-18 (IL-18) upon treatment with NLRP3 stimuli and a reduction in ASC speck formation and oligomerization. Thus, we elucidate a novel regulatory mechanism of the inflammasome signaling pathway mediated by the USP50 deubiquitinating enzyme.
NOD样受体家族蛋白3(NLRP3)介导的炎性小体激活促进半胱天冬酶-1依赖性白细胞介素-1β(IL-1β)的产生,并且需要接头蛋白ASC。与炎性小体信号通路的启动和激活机制相比,控制炎性小体激活的翻译后泛素化/去泛素化机制尚未得到明确阐述。我们在此证明,去泛素化酶USP50与ASC蛋白结合,随后通过去除ASC赖氨酸63连接的多聚泛素化来调节炎性小体信号通路。在人THP-1细胞和小鼠骨髓来源的巨噬细胞中敲低USP50显示,在用NLRP3刺激处理后,前半胱天冬酶-1的切割显著减少,导致IL-1β和白细胞介素-18(IL-18)的分泌减少,以及ASC斑点形成和寡聚化减少。因此,我们阐明了由USP50去泛素化酶介导的炎性小体信号通路的一种新的调节机制。