Wang Qi, Fei Peipei, Gu Hongbo, Zhang Yanmei, Ke Xiaomei, Liu Yuhe
Department of Otolaryngology, Head and Neck Surgery, Peking University First Hospital, Beijing, China.
PLoS One. 2017 Jan 18;12(1):e0170011. doi: 10.1371/journal.pone.0170011. eCollection 2017.
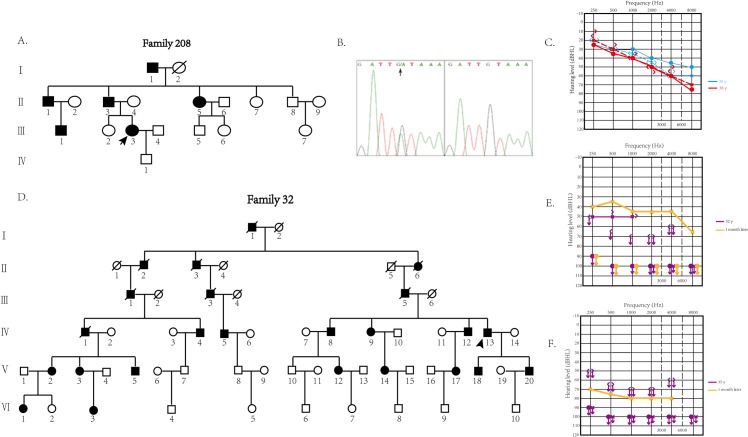
By analyzing the different phenotypes of two Chinese DFNA9 families with the same mutation located in the intervening region between the LCCL and vWFA domains of cochlin and testing the functional changes in the mutant cochlin, we investigated the different pathogeneses for mutations in LCCL and vWFA domains.
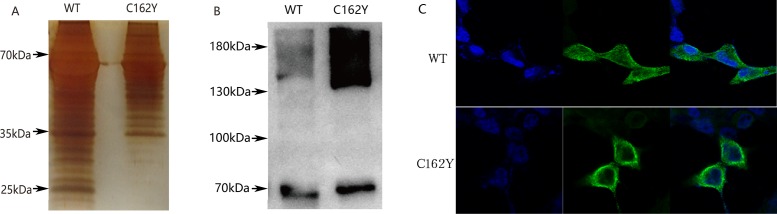
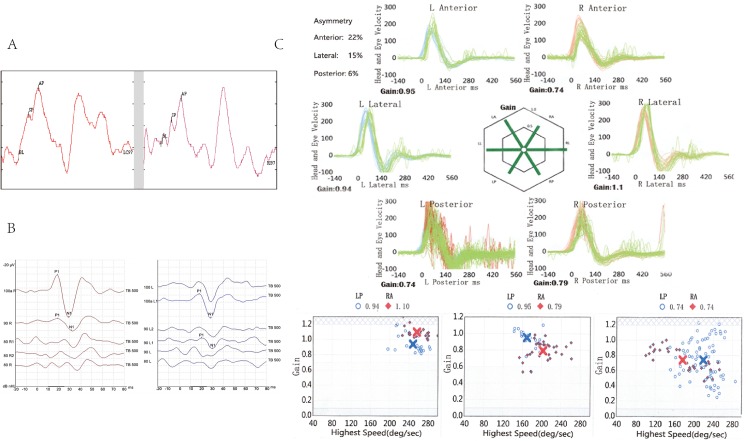
Targeted next-generation sequencing for deafness-related genes was used to identify the mutation in the proband in family #208. The probands of family #208 and family #32 with the same p.C162Y mutation were followed for more than 3 years to evaluate the progression of hearing loss and vestibular dysfunction using pure-tone audiometry, caloric testing, electrocochleogram, vestibular-evoked myogenic potential, and video head-impulse test. The disruption of normal cleavage to produce secreted LCCL domain fragments and the tendency to form aggregations of mutant cochlins were tested by in vitro cell experiments.
The two families showed different clinical symptoms. Family #32 was identified as having early-onset, progressive sensorineural hearing loss, similar to the symptoms in DFNA9 patients with cochlin mutations in the vWFA domain. The proband of family #208 endured late-onset recurrent paroxysmal vertigo attacks and progressively deteriorating hearing, similar to symptoms in those with cochlin mutations in the LCCL domain. We therefore suggest that the disrupted cleavage of the LCCL domain fragment is likely to cause vestibular dysfunction, and aggregation of mutant cochlin caused by mutations in the vWFA domain is responsible for early-onset hearing loss. The p.C162Y mutation causes either disruption of LCCL domain fragment cleavage or aggregation of mutant cochlin, resulting in the different phenotypes in the two families.
This study demonstrates that DFNA9 families with the same genotype may have significantly different phenotypes. The mutation site in cochlin is related to the pathological mechanism underlying the different phenotypes.
通过分析两个中国DFNA9家系的不同表型,这两个家系在耳蜗蛋白的LCCL和vWFA结构域之间的间隔区域存在相同突变,并测试突变型耳蜗蛋白的功能变化,我们研究了LCCL和vWFA结构域突变的不同发病机制。
使用针对耳聋相关基因的靶向二代测序来鉴定208号家系先证者的突变。对208号家系和32号家系具有相同p.C162Y突变的先证者进行了3年多的随访,使用纯音听力测定、冷热试验、耳蜗电图、前庭诱发肌源性电位和视频头脉冲试验来评估听力损失和前庭功能障碍的进展。通过体外细胞实验测试了正常切割产生分泌型LCCL结构域片段的破坏以及突变型耳蜗蛋白形成聚集的倾向。
这两个家系表现出不同的临床症状。32号家系被确定为具有早发性进行性感音神经性听力损失,类似于vWFA结构域中存在耳蜗蛋白突变的DFNA9患者的症状。208号家系的先证者经历了迟发性复发性阵发性眩晕发作和听力逐渐恶化,类似于LCCL结构域中存在耳蜗蛋白突变者的症状。因此,我们认为LCCL结构域片段切割的破坏可能导致前庭功能障碍,而vWFA结构域突变引起的突变型耳蜗蛋白聚集是早发性听力损失的原因。p.C162Y突变导致LCCL结构域片段切割破坏或突变型耳蜗蛋白聚集,导致两个家系出现不同表型。
本研究表明,具有相同基因型的DFNA9家系可能具有显著不同的表型。耳蜗蛋白中的突变位点与不同表型的病理机制有关。