Sanjak Jaleal S, Long Anthony D, Thornton Kevin R
Department of Ecology and Evolutionary Biology, University of California, Irvine, Irvine, California, USA.
Center for Complex Biological Systems, University of California, Irvine, Irvine, California, USA.
PLoS Genet. 2017 Jan 19;13(1):e1006573. doi: 10.1371/journal.pgen.1006573. eCollection 2017 Jan.
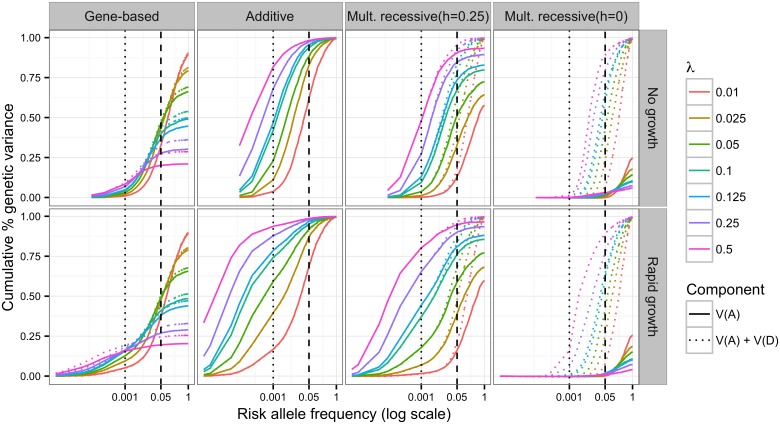
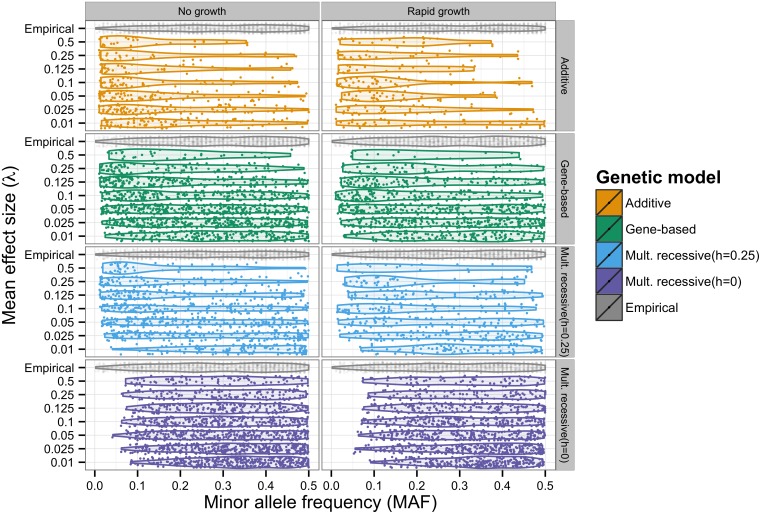
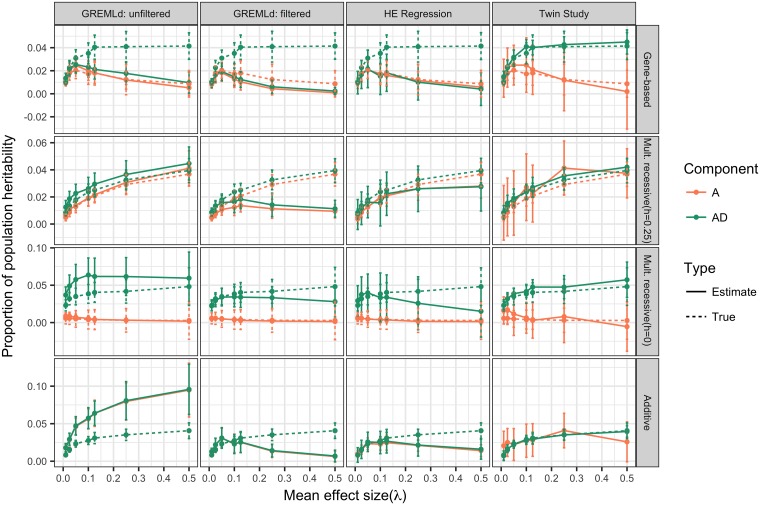
The genetic component of complex disease risk in humans remains largely unexplained. A corollary is that the allelic spectrum of genetic variants contributing to complex disease risk is unknown. Theoretical models that relate population genetic processes to the maintenance of genetic variation for quantitative traits may suggest profitable avenues for future experimental design. Here we use forward simulation to model a genomic region evolving under a balance between recurrent deleterious mutation and Gaussian stabilizing selection. We consider multiple genetic and demographic models, and several different methods for identifying genomic regions harboring variants associated with complex disease risk. We demonstrate that the model of gene action, relating genotype to phenotype, has a qualitative effect on several relevant aspects of the population genetic architecture of a complex trait. In particular, the genetic model impacts genetic variance component partitioning across the allele frequency spectrum and the power of statistical tests. Models with partial recessivity closely match the minor allele frequency distribution of significant hits from empirical genome-wide association studies without requiring homozygous effect sizes to be small. We highlight a particular gene-based model of incomplete recessivity that is appealing from first principles. Under that model, deleterious mutations in a genomic region partially fail to complement one another. This model of gene-based recessivity predicts the empirically observed inconsistency between twin and SNP based estimated of dominance heritability. Furthermore, this model predicts considerable levels of unexplained variance associated with intralocus epistasis. Our results suggest a need for improved statistical tools for region based genetic association and heritability estimation.
人类复杂疾病风险的遗传成分在很大程度上仍未得到解释。一个必然结果是,导致复杂疾病风险的遗传变异的等位基因谱尚不清楚。将群体遗传过程与数量性状遗传变异维持相关联的理论模型,可能为未来的实验设计指明有益的途径。在此,我们使用正向模拟对一个在反复有害突变和高斯稳定选择之间平衡下进化的基因组区域进行建模。我们考虑了多种遗传和人口统计学模型,以及几种不同的方法来识别含有与复杂疾病风险相关变异的基因组区域。我们证明,将基因型与表型联系起来的基因作用模型,对复杂性状群体遗传结构的几个相关方面有定性影响。特别是,遗传模型会影响等位基因频率谱上的遗传方差成分划分以及统计检验的功效。具有部分隐性的模型与经验性全基因组关联研究中显著命中的次要等位基因频率分布紧密匹配,而无需纯合效应大小很小。我们强调了一种基于基因的不完全隐性特殊模型,从第一原理来看很有吸引力。在该模型下,基因组区域中的有害突变部分地无法相互互补。这种基于基因的隐性模型预测了基于双胞胎和单核苷酸多态性(SNP)估计的显性遗传力之间经验性观察到的不一致性。此外,该模型预测了与基因座内上位性相关的相当程度的无法解释的方差。我们的结果表明需要改进用于基于区域的遗传关联和遗传力估计的统计工具。