Balick Daniel J, Do Ron, Cassa Christopher A, Reich David, Sunyaev Shamil R
Division of Genetics, Brigham and Women's Hospital, Boston, Massachusetts, United States of America; Department of Medicine, Harvard Medical School, Boston, Massachusetts, United States of America; Broad Institute of Harvard and MIT, Cambridge, Massachusetts, United States of America.
Broad Institute of Harvard and MIT, Cambridge, Massachusetts, United States of America; The Charles Bronfman Institute for Personalized Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, United States of America; The Center for Statistical Genetics, Icahn School of Medicine at Mount Sinai, New York, New York, United States of America; Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, New York, United States of America.
PLoS Genet. 2015 Aug 28;11(8):e1005436. doi: 10.1371/journal.pgen.1005436. eCollection 2015 Aug.
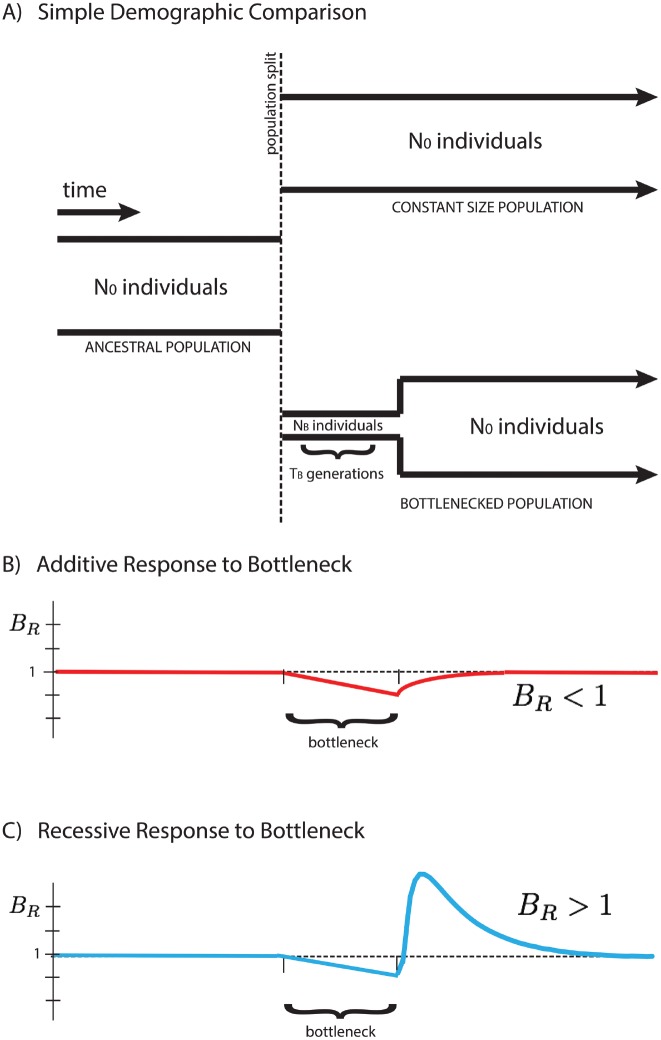
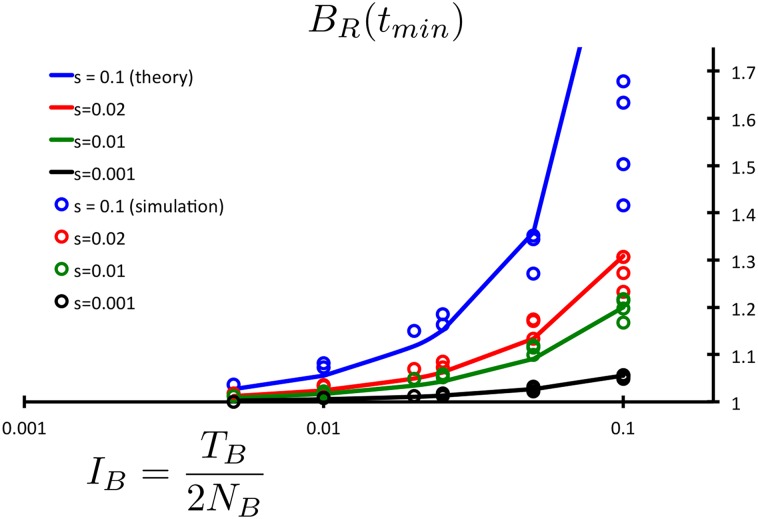
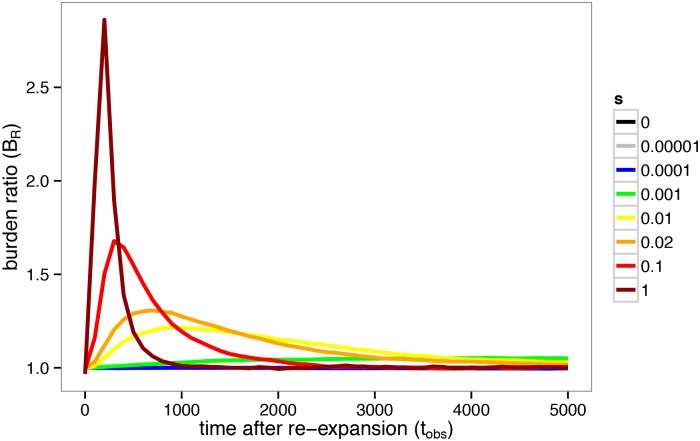
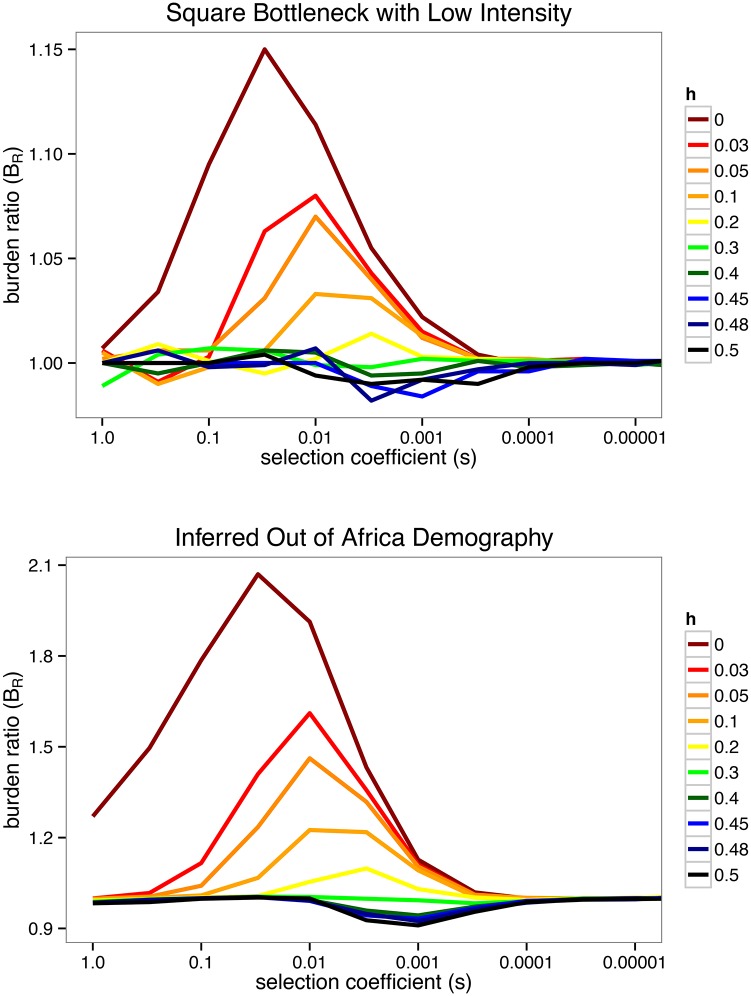
Population bottlenecks followed by re-expansions have been common throughout history of many populations. The response of alleles under selection to such demographic perturbations has been a subject of great interest in population genetics. On the basis of theoretical analysis and computer simulations, we suggest that this response qualitatively depends on dominance. The number of dominant or additive deleterious alleles per haploid genome is expected to be slightly increased following the bottleneck and re-expansion. In contrast, the number of completely or partially recessive alleles should be sharply reduced. Changes of population size expose differences between recessive and additive selection, potentially providing insight into the prevalence of dominance in natural populations. Specifically, we use a simple statistic, [Formula: see text], where xi represents the derived allele frequency, to compare the number of mutations in different populations, and detail its functional dependence on the strength of selection and the intensity of the population bottleneck. We also provide empirical evidence showing that gene sets associated with autosomal recessive disease in humans may have a BR indicative of recessive selection. Together, these theoretical predictions and empirical observations show that complex demographic history may facilitate rather than impede inference of parameters of natural selection.
群体瓶颈继之以重新扩张在许多群体的历史中屡见不鲜。选择作用下的等位基因对这种人口统计学扰动的反应一直是群体遗传学中备受关注的课题。基于理论分析和计算机模拟,我们认为这种反应在性质上取决于显性。每个单倍体基因组中显性或加性有害等位基因的数量在瓶颈和重新扩张之后预计会略有增加。相比之下,完全或部分隐性等位基因的数量应会大幅减少。种群大小的变化揭示了隐性选择和加性选择之间的差异,这可能为了解自然群体中显性的普遍性提供线索。具体而言,我们使用一个简单的统计量[公式:见正文],其中xi代表衍生等位基因频率,来比较不同群体中的突变数量,并详细说明其对选择强度和群体瓶颈强度的功能依赖性。我们还提供了实证证据,表明与人类常染色体隐性疾病相关的基因集可能具有隐性选择的BR特征。这些理论预测和实证观察共同表明,复杂的人口统计学历史可能有助于而非阻碍自然选择参数的推断。