Kapli P, Lutteropp S, Zhang J, Kobert K, Pavlidis P, Stamatakis A, Flouri T
The Exelixis Lab, Scientific Computing Group, Heidelberg Institute for Theoretical Studies, Heidelberg, Germany.
Department of Informatics, Institute of Theoretical Informatics, Karlsruhe Institute of Technology, Karlsruhe, Germany.
Bioinformatics. 2017 Jun 1;33(11):1630-1638. doi: 10.1093/bioinformatics/btx025.
In recent years, molecular species delimitation has become a routine approach for quantifying and classifying biodiversity. Barcoding methods are of particular importance in large-scale surveys as they promote fast species discovery and biodiversity estimates. Among those, distance-based methods are the most common choice as they scale well with large datasets; however, they are sensitive to similarity threshold parameters and they ignore evolutionary relationships. The recently introduced "Poisson Tree Processes" (PTP) method is a phylogeny-aware approach that does not rely on such thresholds. Yet, two weaknesses of PTP impact its accuracy and practicality when applied to large datasets; it does not account for divergent intraspecific variation and is slow for a large number of sequences.
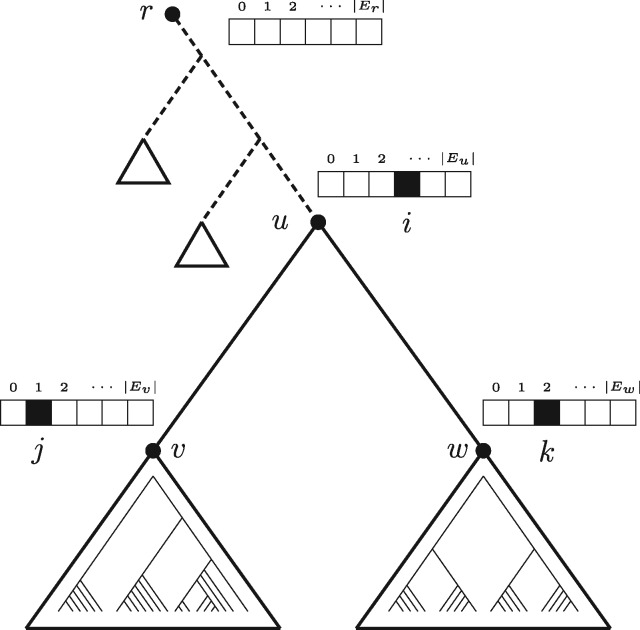
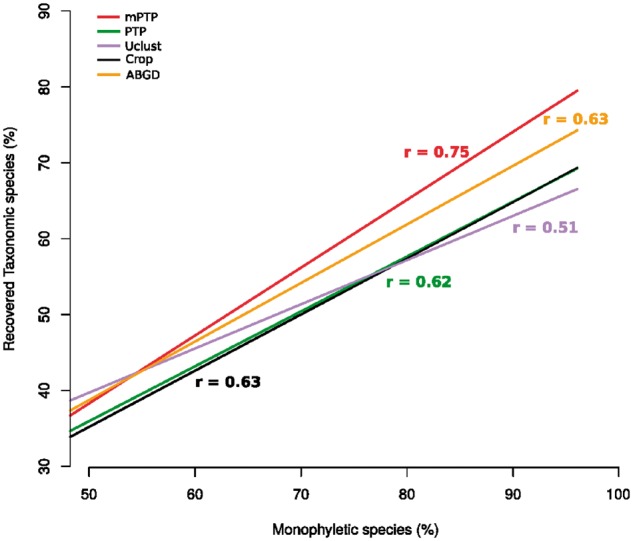
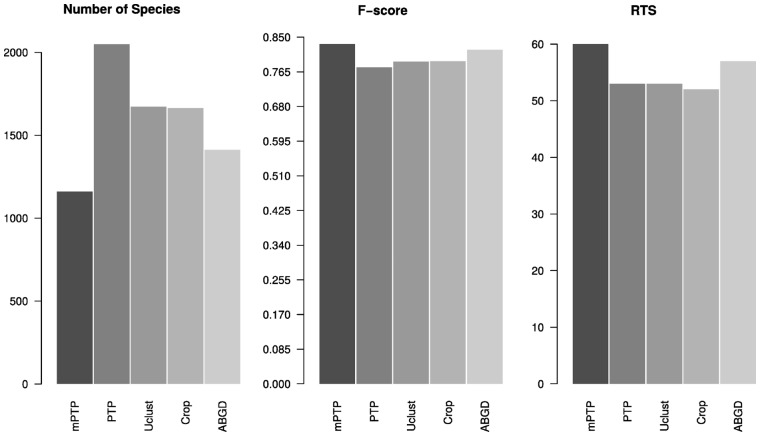
We introduce the multi-rate PTP (mPTP), an improved method that alleviates the theoretical and technical shortcomings of PTP. It incorporates different levels of intraspecific genetic diversity deriving from differences in either the evolutionary history or sampling of each species. Results on empirical data suggest that mPTP is superior to PTP and popular distance-based methods as it, consistently yields more accurate delimitations with respect to the taxonomy (i.e., identifies more taxonomic species, infers species numbers closer to the taxonomy). Moreover, mPTP does not require any similarity threshold as input. The novel dynamic programming algorithm attains a speedup of at least five orders of magnitude compared to PTP, allowing it to delimit species in large (meta-) barcoding data. In addition, Markov Chain Monte Carlo sampling provides a comprehensive evaluation of the inferred delimitation in just a few seconds for millions of steps, independently of tree size.
mPTP is implemented in C and is available for download at http://github.com/Pas-Kapli/mptp under the GNU Affero 3 license. A web-service is available at http://mptp.h-its.org .
: paschalia.kapli@h-its.org or alexandros.stamatakis@h-its.org or tomas.flouri@h-its.org.
Supplementary data are available at Bioinformatics online.
近年来,分子物种界定已成为量化和分类生物多样性的常规方法。条形码方法在大规模调查中尤为重要,因为它们有助于快速发现物种并估计生物多样性。其中,基于距离的方法是最常见的选择,因为它们在处理大型数据集时扩展性良好;然而,它们对相似性阈值参数敏感,并且忽略了进化关系。最近引入的“泊松树过程”(PTP)方法是一种系统发育感知方法,不依赖于此类阈值。然而,PTP的两个弱点在应用于大型数据集时会影响其准确性和实用性;它没有考虑种内差异,并且对于大量序列来说速度较慢。
我们引入了多速率PTP(mPTP),这是一种改进方法,可缓解PTP的理论和技术缺陷。它纳入了因每个物种的进化历史或采样差异而产生的不同水平的种内遗传多样性。实证数据结果表明,mPTP优于PTP和流行的基于距离的方法,因为它始终能在分类学方面产生更准确的界定(即识别出更多分类学物种,推断出更接近分类学的物种数量)。此外,mPTP不需要任何相似性阈值作为输入。与PTP相比时,新颖的动态规划算法实现了至少五个数量级的加速,使其能够在大型(元)条形码数据中界定物种。此外,马尔可夫链蒙特卡罗采样仅需几秒钟就能对推断的界定进行数百万步的全面评估,且与树的大小无关。
mPTP用C语言实现,可在http://github.com/Pas-Kapli/mptp上根据GNU Affero 3许可下载。可在http://mptp.h-its.org上使用网络服务。
paschalia.kapli@h-its.org或alexandros.stamatakis@h-its.org或tomas.flouri@h-its.org。
补充数据可在《生物信息学》在线获取。