Doungpan Narumol, Engchuan Worrawat, Chan Jonathan H, Meechai Asawin
Biological Engineering Program, Faculty of Engineering, King Mongkut's University of Technology Thonburi, Bangkok, Thailand.
The Centre for Applied Genomics, Genetics and Genome Biology, The Hospital for Sick Children, Toronto, ON, Canada.
BMC Med Genomics. 2016 Dec 5;9(Suppl 3):70. doi: 10.1186/s12920-016-0231-4.
Gene expression has been used to identify disease gene biomarkers, but there are ongoing challenges. Single gene or gene-set biomarkers are inadequate to provide sufficient understanding of complex disease mechanisms and the relationship among those genes. Network-based methods have thus been considered for inferring the interaction within a group of genes to further study the disease mechanism. Recently, the Gene-Network-based Feature Set (GNFS), which is capable of handling case-control and multiclass expression for gene biomarker identification, has been proposed, partly taking into account of network topology. However, its performance relies on a greedy search for building subnetworks and thus requires further improvement. In this work, we establish a new approach named Gene Sub-Network-based Feature Selection (GSNFS) by implementing the GNFS framework with two proposed searching and scoring algorithms, namely gene-set-based (GS) search and parent-node-based (PN) search, to identify subnetworks. An additional dataset is used to validate the results.
The two proposed searching algorithms of the GSNFS method for subnetwork expansion are concerned with the degree of connectivity and the scoring scheme for building subnetworks and their topology. For each iteration of expansion, the neighbour genes of a current subnetwork, whose expression data improved the overall subnetwork score, is recruited. While the GS search calculated the subnetwork score using an activity score of a current subnetwork and the gene expression values of its neighbours, the PN search uses the expression value of the corresponding parent of each neighbour gene. Four lung cancer expression datasets were used for subnetwork identification. In addition, using pathway data and protein-protein interaction as network data in order to consider the interaction among significant genes were discussed. Classification was performed to compare the performance of the identified gene subnetworks with three subnetwork identification algorithms.
The two searching algorithms resulted in better classification and gene/gene-set agreement compared to the original greedy search of the GNFS method. The identified lung cancer subnetwork using the proposed searching algorithm resulted in an improvement of the cross-dataset validation and an increase in the consistency of findings between two independent datasets. The homogeneity measurement of the datasets was conducted to assess dataset compatibility in cross-dataset validation. The lung cancer dataset with higher homogeneity showed a better result when using the GS search while the dataset with low homogeneity showed a better result when using the PN search. The 10-fold cross-dataset validation on the independent lung cancer datasets showed higher classification performance of the proposed algorithms when compared with the greedy search in the original GNFS method.
The proposed searching algorithms provide a higher number of genes in the subnetwork expansion step than the greedy algorithm. As a result, the performance of the subnetworks identified from the GSNFS method was improved in terms of classification performance and gene/gene-set level agreement depending on the homogeneity of the datasets used in the analysis. Some common genes obtained from the four datasets using different searching algorithms are genes known to play a role in lung cancer. The improvement of classification performance and the gene/gene-set level agreement, and the biological relevance indicated the effectiveness of the GSNFS method for gene subnetwork identification using expression data.
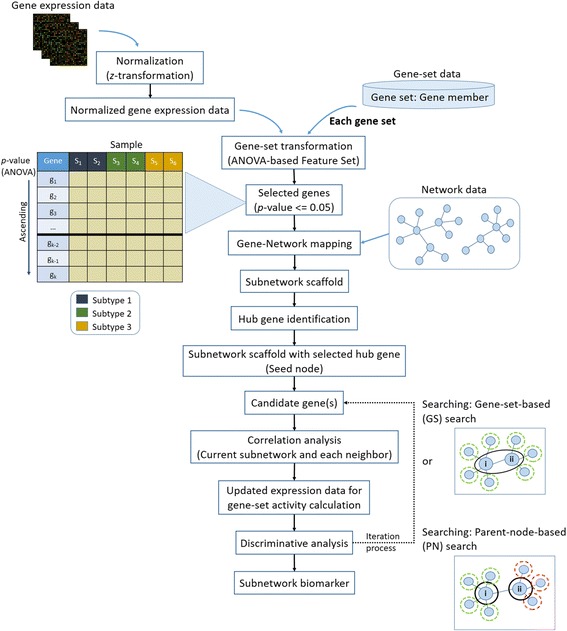
基因表达已被用于识别疾病基因生物标志物,但仍存在诸多挑战。单基因或基因集生物标志物不足以充分理解复杂的疾病机制以及这些基因之间的关系。因此,基于网络的方法被用于推断一组基因内的相互作用,以进一步研究疾病机制。最近,有人提出了基于基因网络的特征集(GNFS),它能够处理病例对照和多类基因表达以进行基因生物标志物识别,部分考虑了网络拓扑结构。然而,其性能依赖于构建子网的贪婪搜索,因此需要进一步改进。在这项工作中,我们通过用两种提出的搜索和评分算法,即基于基因集(GS)的搜索和基于父节点(PN)的搜索,来实现GNFS框架,建立了一种名为基于基因子网的特征选择(GSNFS)的新方法,以识别子网。使用一个额外的数据集来验证结果。
GSNFS方法用于子网扩展的两种提出的搜索算法涉及连通度以及构建子网及其拓扑结构的评分方案。对于每次扩展迭代,招募当前子网的邻居基因,其表达数据改善了整个子网的分数。虽然GS搜索使用当前子网的活性分数及其邻居的基因表达值来计算子网分数,但PN搜索使用每个邻居基因相应父节点的表达值。使用四个肺癌表达数据集进行子网识别。此外,还讨论了使用通路数据和蛋白质 - 蛋白质相互作用作为网络数据,以便考虑显著基因之间的相互作用。进行分类以将识别出的基因子网的性能与三种子网识别算法进行比较。
与GNFS方法的原始贪婪搜索相比,这两种搜索算法在分类以及基因/基因集一致性方面表现更好。使用提出的搜索算法识别出的肺癌子网在跨数据集验证方面有所改进,并且两个独立数据集之间的发现一致性有所增加。对数据集进行同质性测量以评估跨数据集验证中的数据集兼容性。同质性较高的肺癌数据集在使用GS搜索时显示出更好的结果,而同质性较低的数据集在使用PN搜索时显示出更好的结果。在独立的肺癌数据集上进行的10倍交叉数据集验证表明,与原始GNFS方法中的贪婪搜索相比,所提出的算法具有更高的分类性能。
所提出的搜索算法在子网扩展步骤中比贪婪算法提供了更多的基因。因此,根据分析中使用的数据集的同质性,从GSNFS方法识别出的子网在分类性能和基因/基因集水平一致性方面得到了改进。使用不同搜索算法从四个数据集中获得的一些常见基因是已知在肺癌中起作用的基因。分类性能和基因/基因集水平一致性的提高以及生物学相关性表明了GSNFS方法使用表达数据进行基因子网识别的有效性。