Department of Computer Science and Engineering, University of Nevada, Reno, 89557, Nevada, United States.
Wayne State University, Department of Computer Science, Detroit, 48202, Michigan, United States.
Sci Rep. 2020 Mar 6;10(1):4188. doi: 10.1038/s41598-020-60981-9.
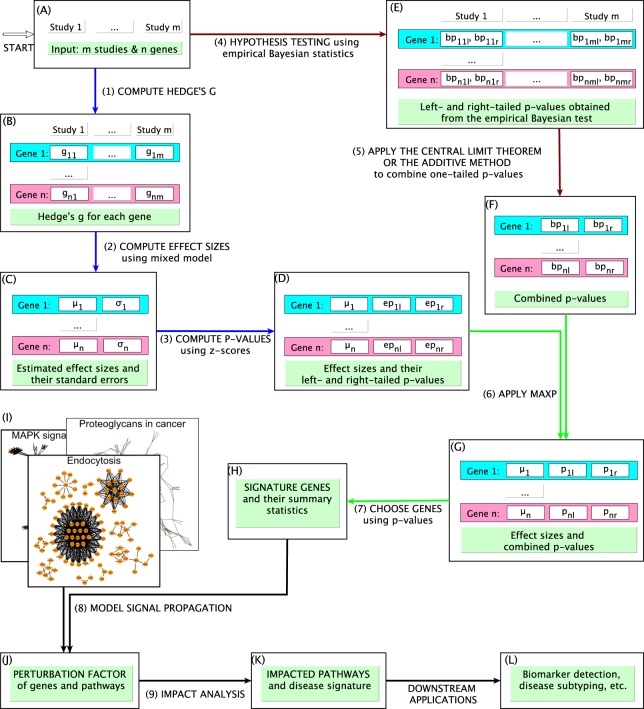
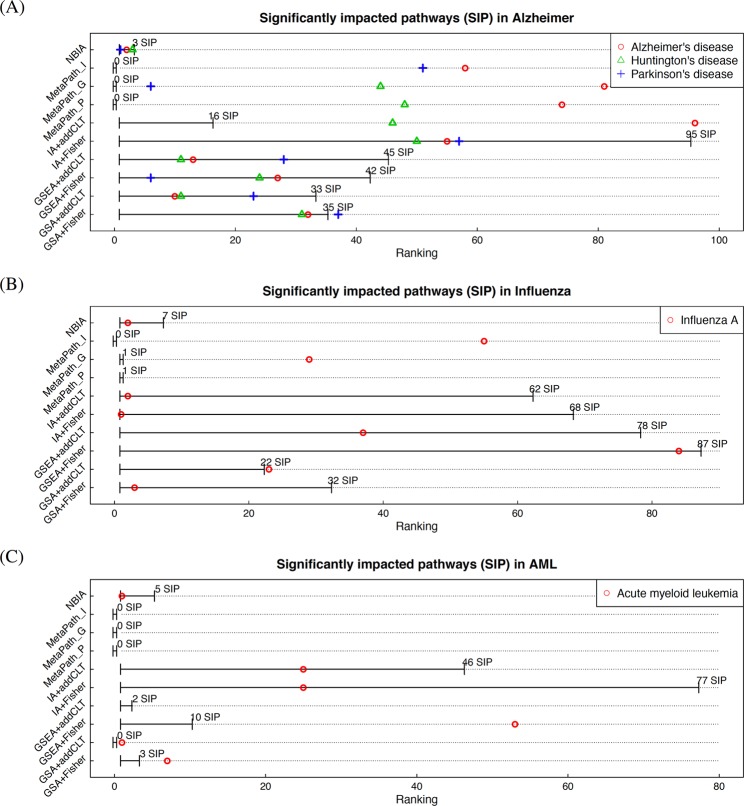
With the explosion of high-throughput data, effective integrative analyses are needed to decipher the knowledge accumulated in biological databases. Existing meta-analysis approaches in systems biology often focus on hypothesis testing and neglect real expression changes, i.e. effect sizes, across independent studies. In addition, most integrative tools completely ignore the topological order of gene regulatory networks that hold key characteristics in understanding biological processes. Here we introduce a novel meta-analysis framework, Network-Based Integrative Analysis (NBIA), that transforms the challenging meta-analysis problem into a set of standard pathway analysis problems that have been solved efficiently. NBIA utilizes techniques from classical and modern meta-analysis, as well as a network-based analysis, in order to identify patterns of genes and networks that are consistently impacted across multiple studies. We assess the performance of NBIA by comparing it with nine meta-analysis approaches: Impact Analysis, GSA, and GSEA combined with classical meta-analysis methods (Fisher's and the additive method), plus the three MetaPath approaches that employ multiple datasets. The 10 approaches have been tested on 1,737 samples from 27 expression datasets related to Alzheimer's disease, acute myeloid leukemia (AML), and influenza. For all of the three diseases, NBIA consistently identifies biological pathways relevant to the underlying diseases while the other 9 methods fail to capture the key phenomena. The identified AML signature is also validated on a completely independent cohort of 167 AML patients. In this independent cohort, the proposed signature identifies two groups of patients that have significantly different survival profiles (Cox p-value 2 × 10). The NBIA framework will be included in the next release of BLMA Bioconductor package (http://bioconductor.org/packages/release/bioc/html/BLMA.html).
随着高通量数据的爆炸式增长,需要有效的综合分析来破译生物数据库中积累的知识。系统生物学中现有的荟萃分析方法通常侧重于假设检验,而忽略了独立研究中真实的表达变化,即效应大小。此外,大多数综合工具完全忽略了基因调控网络的拓扑顺序,而这些网络在理解生物过程方面具有关键特征。在这里,我们引入了一种新的荟萃分析框架,即基于网络的综合分析(NBIA),它将具有挑战性的荟萃分析问题转化为一组已经得到有效解决的标准途径分析问题。NBIA 利用经典荟萃分析和现代荟萃分析技术以及基于网络的分析技术,以识别在多个研究中一致受到影响的基因和网络模式。我们通过将 NBIA 与九种荟萃分析方法进行比较来评估其性能:影响分析、GSA 以及与经典荟萃分析方法(Fisher 和加法方法)结合的 GSEA,再加上采用多个数据集的三种 MetaPath 方法。这 10 种方法已经在 27 个与阿尔茨海默病、急性髓系白血病(AML)和流感相关的表达数据集的 1737 个样本上进行了测试。对于所有三种疾病,NBIA 始终能够识别与潜在疾病相关的生物学途径,而其他 9 种方法则无法捕捉到关键现象。所识别的 AML 特征也在一个完全独立的 167 个 AML 患者队列上进行了验证。在这个独立的队列中,所提出的特征将患者分为两组,这两组患者的生存情况有显著差异(Cox p 值为 2×10)。NBIA 框架将包含在下一个 BLMA Bioconductor 包的版本中(http://bioconductor.org/packages/release/bioc/html/BLMA.html)。