Cooper Helen M, Yang Yang, Ylikallio Emil, Khairullin Rafil, Woldegebriel Rosa, Lin Kai-Lan, Euro Liliya, Palin Eino, Wolf Alexander, Trokovic Ras, Isohanni Pirjo, Kaakkola Seppo, Auranen Mari, Lönnqvist Tuula, Wanrooij Sjoerd, Tyynismaa Henna
Åbo Akademi University, Faculty of Natural Sciences and Technology, Turku, Finland.
Research Programs Unit, Molecular Neurology, University of Helsinki, Helsinki, Finland.
Hum Mol Genet. 2017 Apr 15;26(8):1432-1443. doi: 10.1093/hmg/ddx042.
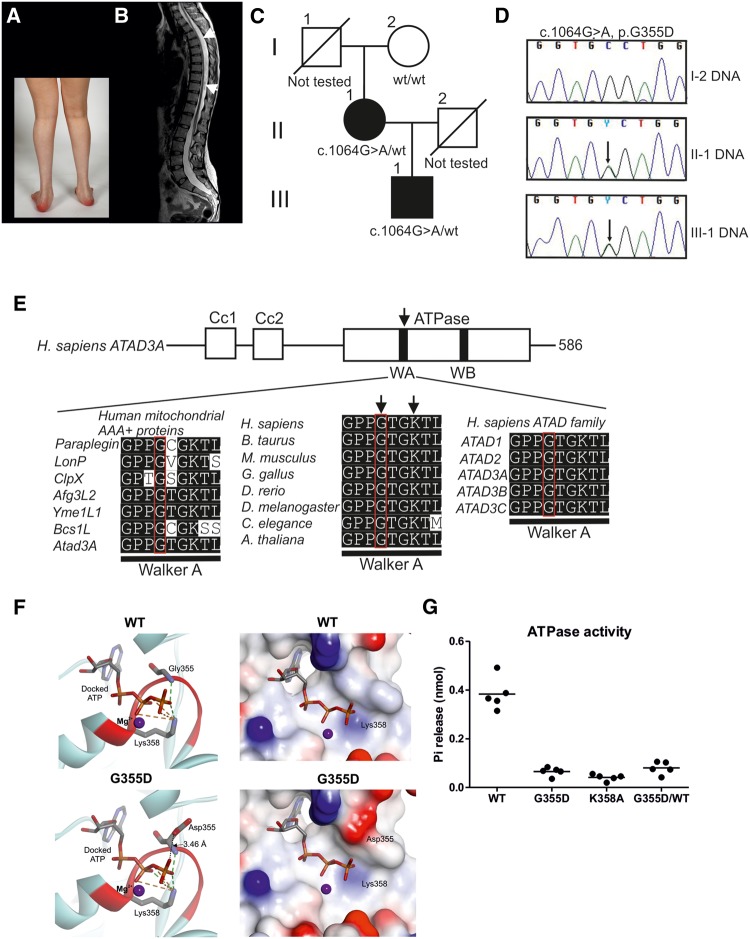
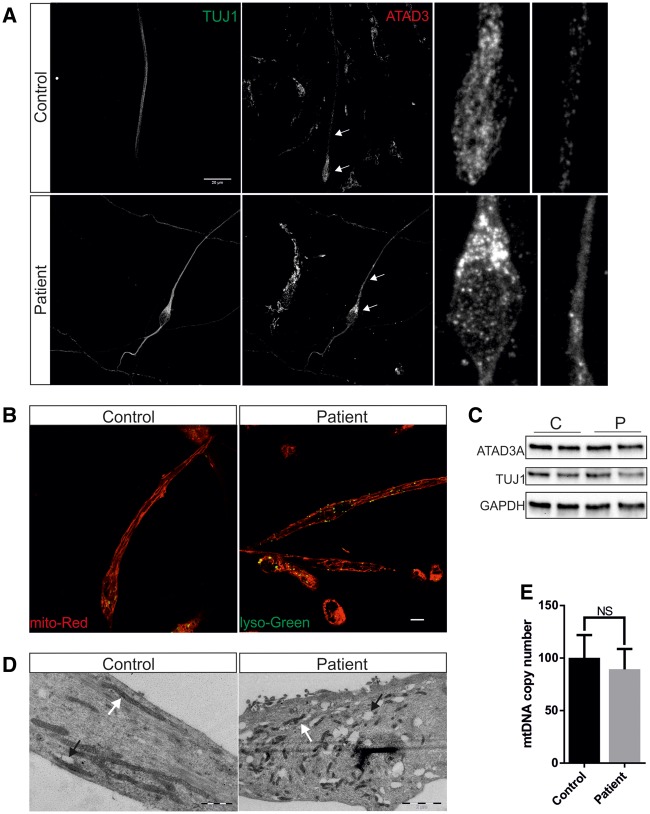
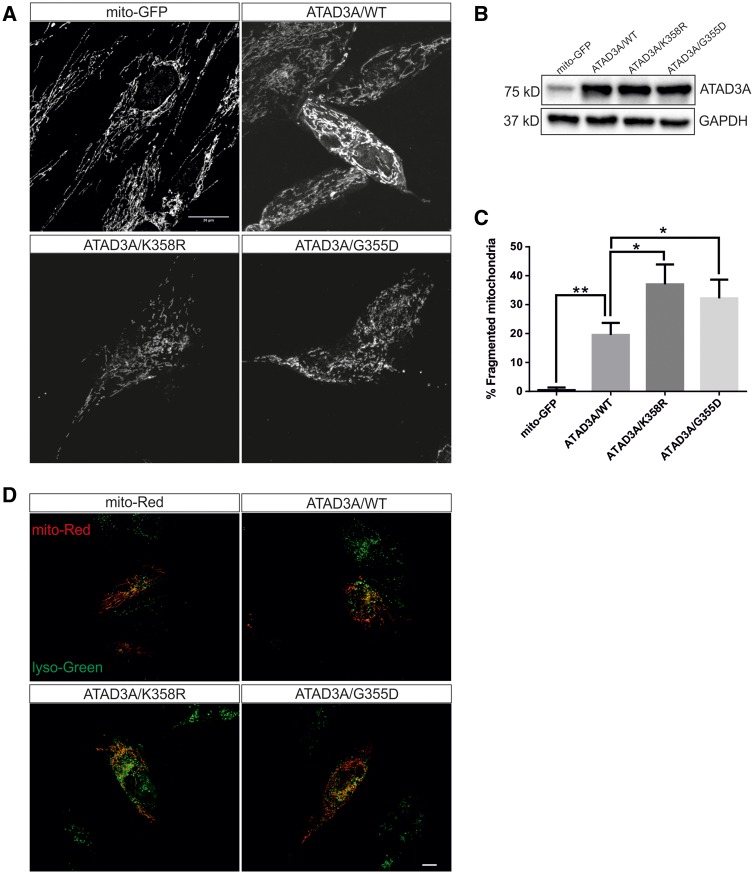
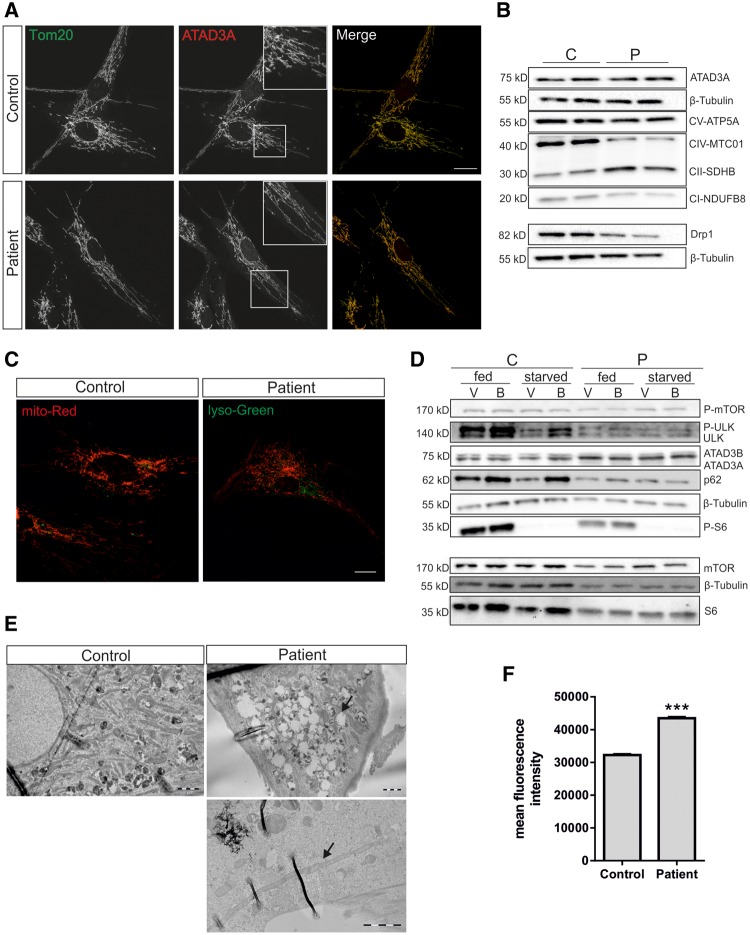
De novo mutations in ATAD3A (ATPase family AAA-domain containing protein 3A) were recently found to cause a neurological syndrome with developmental delay, hypotonia, spasticity, optic atrophy, axonal neuropathy, and hypertrophic cardiomyopathy. Using whole-exome sequencing, we identified a dominantly inherited heterozygous variant c.1064G > A (p.G355D) in ATAD3A in a mother presenting with hereditary spastic paraplegia (HSP) and axonal neuropathy and her son with dyskinetic cerebral palsy, both with disease onset in childhood. HSP is a clinically and genetically heterogeneous disorder of the upper motor neurons. Symptoms beginning in early childhood may resemble spastic cerebral palsy. The function of ATAD3A, a mitochondrial inner membrane AAA ATPase, is yet undefined. AAA ATPases form hexameric rings, which are catalytically dependent on the co-operation of the subunits. The dominant-negative patient mutation affects the Walker A motif, which is responsible for ATP binding in the AAA module of ATAD3A, and we show that the recombinant mutant ATAD3A protein has a markedly reduced ATPase activity. We further show that overexpression of the mutant ATAD3A fragments the mitochondrial network and induces lysosome mass. Similarly, we observed altered dynamics of the mitochondrial network and increased lysosomes in patient fibroblasts and neurons derived through differentiation of patient-specific induced pluripotent stem cells. These alterations were verified in patient fibroblasts to associate with upregulated basal autophagy through mTOR inactivation, resembling starvation. Mutations in ATAD3A can thus be dominantly inherited and underlie variable neurological phenotypes, including HSP, with intrafamiliar variability. This finding extends the group of mitochondrial inner membrane AAA proteins associated with spasticity.
最近发现,ATAD3A(含ATP酶家族AAA结构域蛋白3A)的新生突变会导致一种神经综合征,伴有发育迟缓、肌张力减退、痉挛、视神经萎缩、轴索性神经病和肥厚型心肌病。通过全外显子组测序,我们在一位患有遗传性痉挛性截瘫(HSP)和轴索性神经病的母亲及其患有运动障碍型脑瘫的儿子中,发现了ATAD3A基因中一个显性遗传的杂合变异c.1064G>A(p.G355D),两人均在儿童期发病。HSP是一种临床和遗传异质性的上运动神经元疾病。儿童早期出现的症状可能类似于痉挛性脑瘫。ATAD3A是一种线粒体内膜AAA型ATP酶,其功能尚不清楚。AAA型ATP酶形成六聚体环,其催化作用依赖于亚基的协同作用。显性负性的患者突变影响沃克A基序,该基序负责ATAD3A的AAA模块中的ATP结合,我们发现重组突变型ATAD3A蛋白的ATP酶活性明显降低。我们进一步发现,突变型ATAD3A的过表达会使线粒体网络碎片化并诱导溶酶体增多。同样,我们在患者成纤维细胞以及通过患者特异性诱导多能干细胞分化得到的神经元中,观察到线粒体网络动态改变和溶酶体增多。这些改变在患者成纤维细胞中得到证实,与通过mTOR失活上调基础自噬有关,类似于饥饿状态。因此,ATAD3A突变可显性遗传,并成为包括HSP在内的多种神经表型的基础,存在家族内变异性。这一发现扩展了与痉挛相关的线粒体内膜AAA蛋白组。