Aging and Metabolism Research Program, Oklahoma Medical Research Foundation, Oklahoma City, OK, 73104, USA.
Institute of Human Genetics, Technical University Munich, Munich, Germany.
Genome Med. 2021 Apr 12;13(1):55. doi: 10.1186/s13073-021-00873-3.
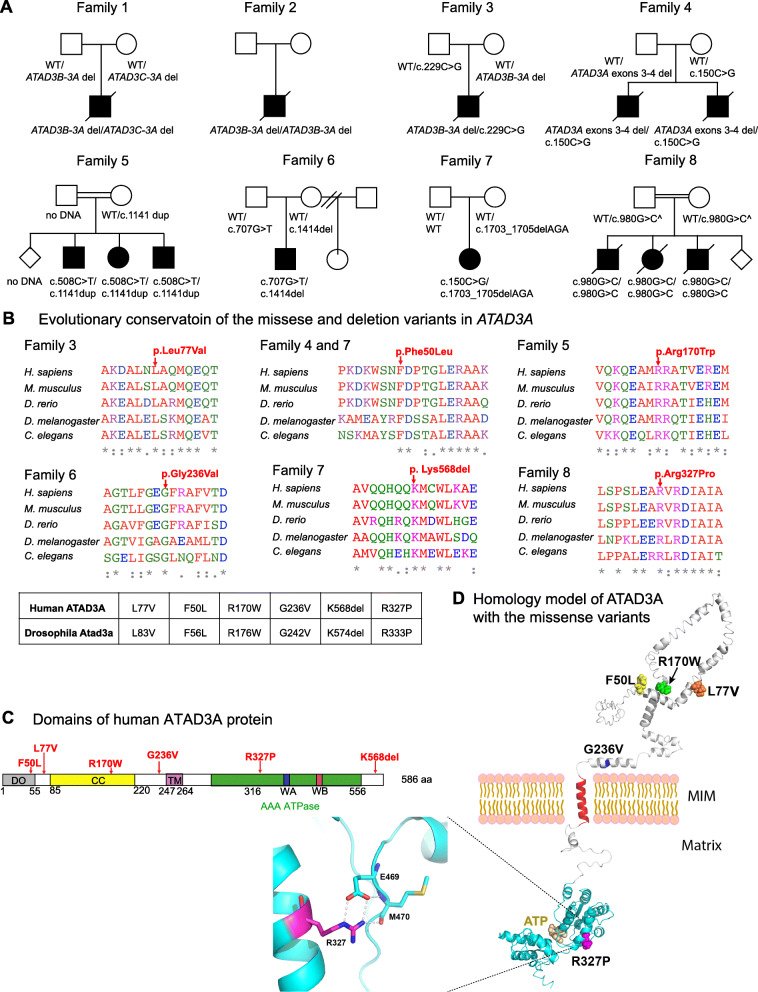
ATPase family AAA-domain containing protein 3A (ATAD3A) is a nuclear-encoded mitochondrial membrane-anchored protein involved in diverse processes including mitochondrial dynamics, mitochondrial DNA organization, and cholesterol metabolism. Biallelic deletions (null), recessive missense variants (hypomorph), and heterozygous missense variants or duplications (antimorph) in ATAD3A lead to neurological syndromes in humans.
To expand the mutational spectrum of ATAD3A variants and to provide functional interpretation of missense alleles in trans to deletion alleles, we performed exome sequencing for identification of single nucleotide variants (SNVs) and copy number variants (CNVs) in ATAD3A in individuals with neurological and mitochondrial phenotypes. A Drosophila Atad3a Gal4 knockin-null allele was generated using CRISPR-Cas9 genome editing technology to aid the interpretation of variants.
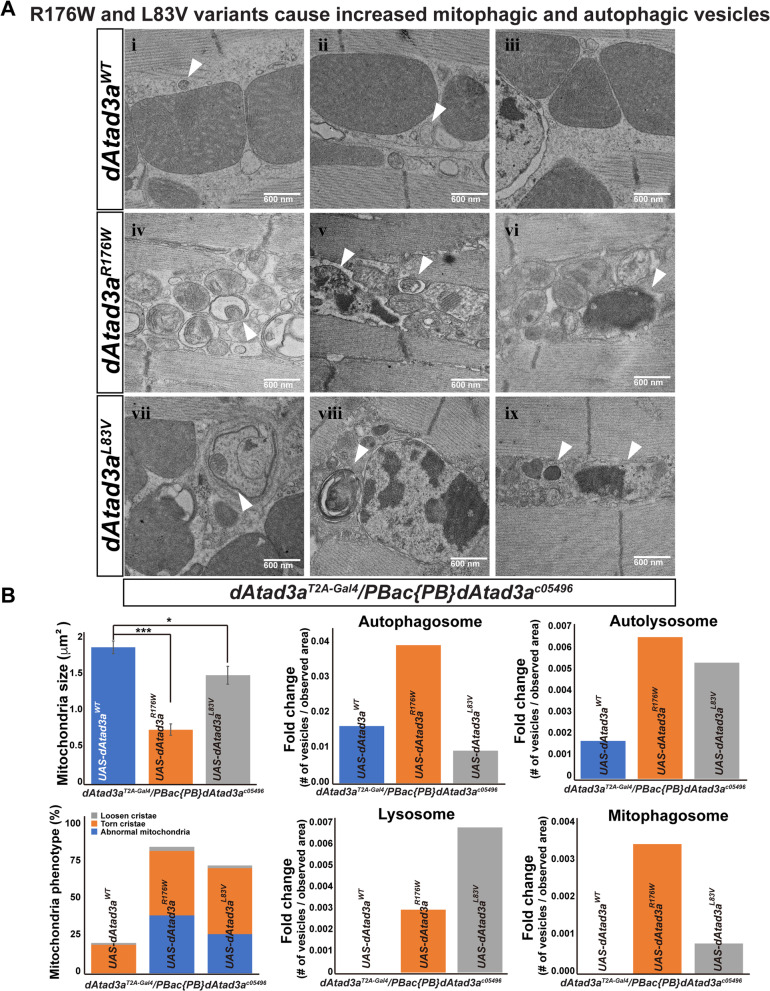
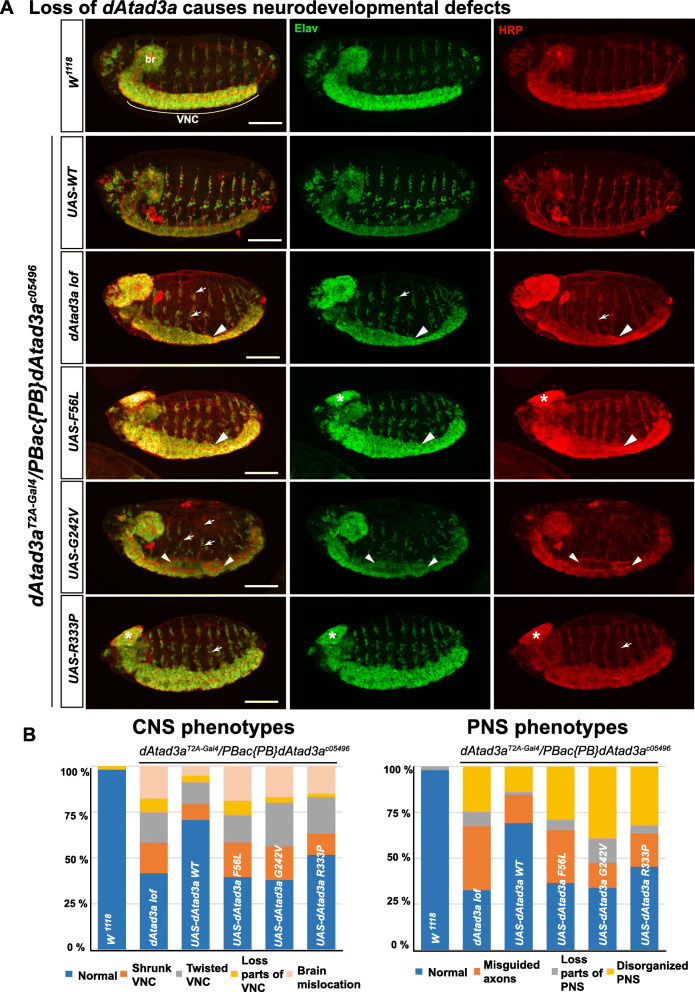
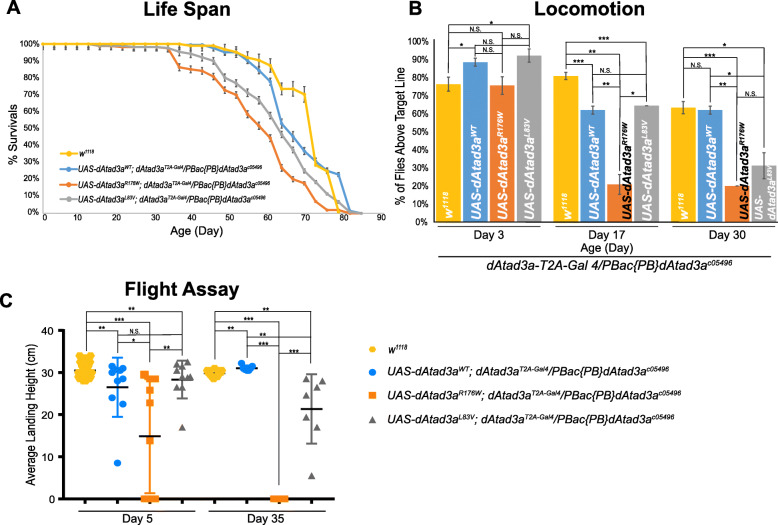
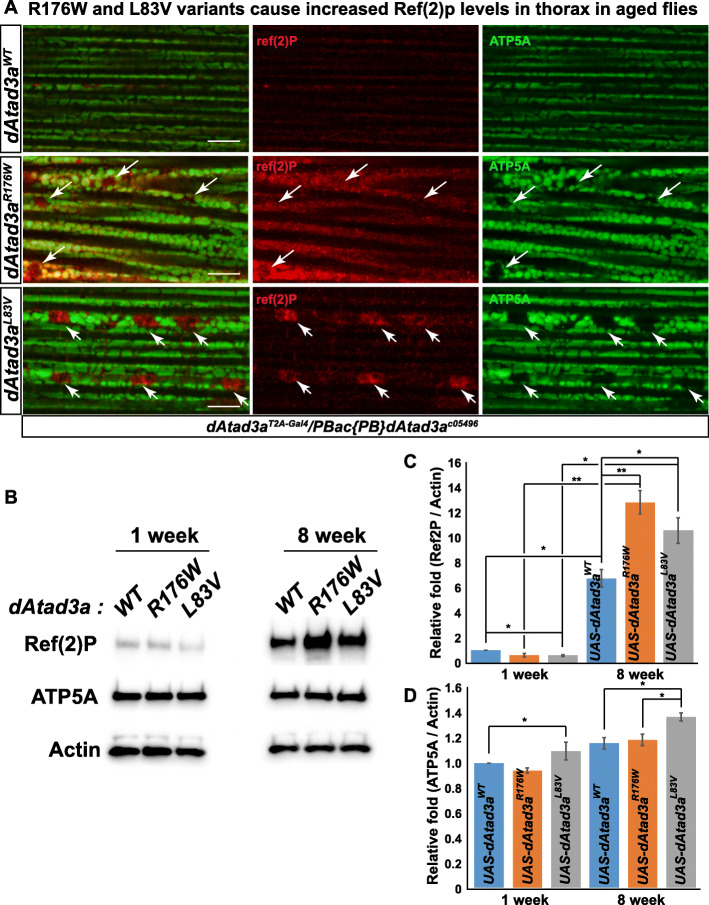
We report 13 individuals from 8 unrelated families with biallelic ATAD3A variants. The variants included four missense variants inherited in trans to loss-of-function alleles (p.(Leu77Val), p.(Phe50Leu), p.(Arg170Trp), p.(Gly236Val)), a homozygous missense variant p.(Arg327Pro), and a heterozygous non-frameshift indel p.(Lys568del). Affected individuals exhibited findings previously associated with ATAD3A pathogenic variation, including developmental delay, hypotonia, congenital cataracts, hypertrophic cardiomyopathy, and cerebellar atrophy. Drosophila studies indicated that Phe50Leu, Gly236Val, Arg327Pro, and Lys568del are severe loss-of-function alleles leading to early developmental lethality. Further, we showed that Phe50Leu, Gly236Val, and Arg327Pro cause neurogenesis defects. On the contrary, Leu77Val and Arg170Trp are partial loss-of-function alleles that cause progressive locomotion defects and whose expression leads to an increase in autophagy and mitophagy in adult muscles.
Our findings expand the allelic spectrum of ATAD3A variants and exemplify the use of a functional assay in Drosophila to aid variant interpretation.
ATP 酶家族 AAA 结构域包含蛋白 3A(ATAD3A)是一种核编码的线粒体膜锚定蛋白,参与多种过程,包括线粒体动力学、线粒体 DNA 组织和胆固醇代谢。ATAD3A 的双等位基因缺失(无功能)、隐性错义变异(功能减弱)和杂合错义变异或重复(功能拮抗)导致人类出现神经综合征。
为了扩展 ATAD3A 变异的突变谱,并为显性缺失等位基因的错义等位基因提供功能解释,我们对具有神经和线粒体表型的个体进行了 ATAD3A 的外显子组测序,以鉴定单核苷酸变异(SNVs)和拷贝数变异(CNVs)。使用 CRISPR-Cas9 基因组编辑技术生成了一个果蝇 Atad3a Gal4 敲入缺失等位基因,以帮助解释变异。
我们报告了 8 个无关家族的 13 名个体存在双等位基因 ATAD3A 变异。这些变异包括 4 个错义变异,这些变异是在显性缺失等位基因的背景下遗传的(p.(Leu77Val), p.(Phe50Leu), p.(Arg170Trp), p.(Gly236Val)),一个纯合的错义变异 p.(Arg327Pro)和一个杂合的非移码缺失 p.(Lys568del)。受影响的个体表现出与 ATAD3A 致病性变异相关的先前发现,包括发育迟缓、张力减退、先天性白内障、肥厚型心肌病和小脑萎缩。果蝇研究表明,Phe50Leu、Gly236Val、Arg327Pro 和 Lys568del 是导致早期发育致死的严重功能丧失等位基因。此外,我们还表明 Phe50Leu、Gly236Val 和 Arg327Pro 导致神经发生缺陷。相反,Leu77Val 和 Arg170Trp 是部分功能丧失等位基因,导致进行性运动缺陷,其表达导致成年肌肉中自噬和线粒体自噬增加。
我们的发现扩展了 ATAD3A 变异的等位基因谱,并举例说明了使用果蝇中的功能测定来辅助变异解释。