Xu Lingyang, Haasl Ryan J, Sun Jiajie, Zhou Yang, Bickhart Derek M, Li Junya, Song Jiuzhou, Sonstegard Tad S, Van Tassell Curtis P, Lewin Harris A, Liu George E
Animal Genomics and Improvement Laboratory, Agricultural Research Service, Beltsville, MD.
Institute of Animal Science, Chinese Academy of Agricultural Sciences, Beijing, China.
Genome Biol Evol. 2017 Jan 1;9(1):20-31. doi: 10.1093/gbe/evw256.
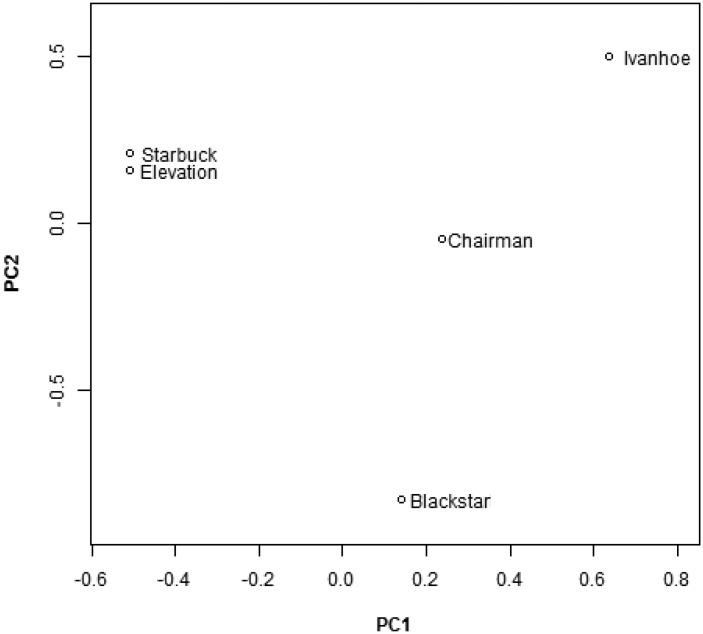
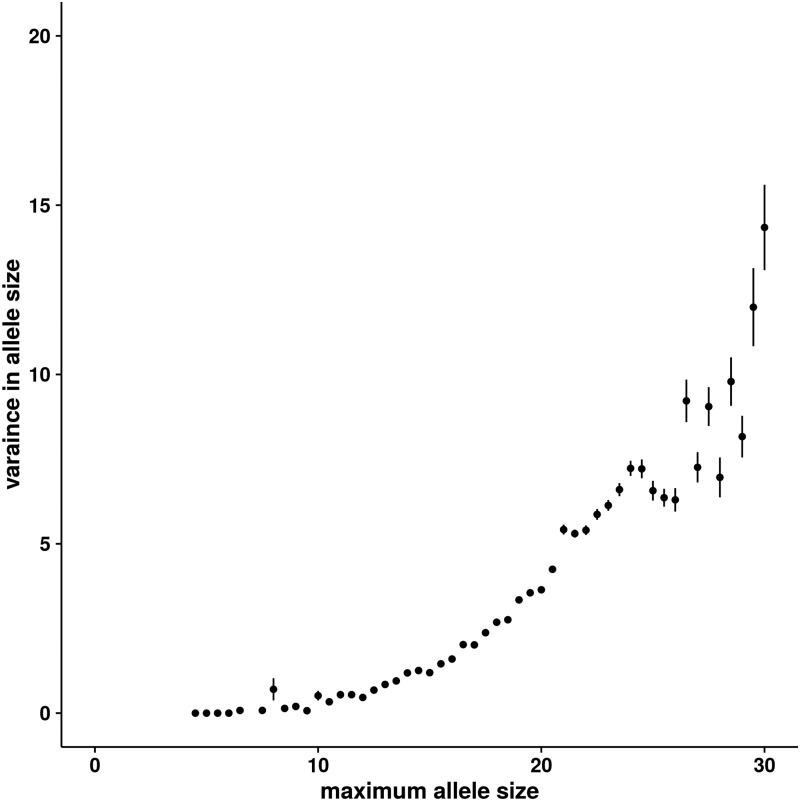
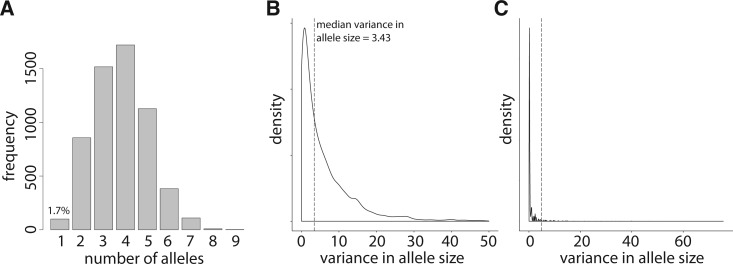
Short tandem repeats (STRs), or microsatellites, are genetic variants with repetitive 2–6 base pair motifs in many mammalian genomes. Using high-throughput sequencing and experimental validations, we systematically profiled STRs in five Holsteins. We identified a total of 60,106 microsatellites and generated the first high-resolution STR map, representing a substantial pool of polymorphism in dairy cattle. We observed significant STRs overlap with functional genes and quantitative trait loci (QTL). We performed evolutionary and population genetic analyses using over 20,000 common dinucleotide STRs. Besides corroborating the well-established positive correlation between allele size and variance in allele size, these analyses also identified dozens of outlier STRs based on two anomalous relationships that counter expected characteristics of neutral evolution. And one STR locus overlaps with a significant region of a summary statistic designed to detect STR-related selection. Additionally, our results showed that only 57.1% of STRs located within SNP-based linkage disequilibrium (LD) blocks whereas the other 42.9% were out of blocks. Therefore, a substantial number of STRs are not tagged by SNPs in the cattle genome, likely due to STR's distinct mutation mechanism and elevated polymorphism. This study provides the foundation for future STR-based studies of cattle genome evolution and selection.
短串联重复序列(STRs),即微卫星,是许多哺乳动物基因组中具有2 - 6个碱基对重复基序的遗传变异。通过高通量测序和实验验证,我们系统地分析了五头荷斯坦奶牛的STRs。我们总共鉴定出60,106个微卫星,并生成了首张高分辨率STR图谱,这代表了奶牛中大量的多态性库。我们观察到显著的STRs与功能基因和数量性状位点(QTL)重叠。我们使用超过20,000个常见的二核苷酸STRs进行了进化和群体遗传学分析。除了证实等位基因大小与等位基因大小方差之间已确立的正相关外,这些分析还基于两种与中性进化预期特征相悖的异常关系,鉴定出了数十个异常STRs。并且一个STR位点与一个旨在检测STR相关选择的汇总统计量的显著区域重叠。此外,我们的结果表明,只有57.1%的STRs位于基于单核苷酸多态性(SNP)的连锁不平衡(LD)块内,而另外42.9%不在块内。因此,牛基因组中大量的STRs未被SNP标记,这可能是由于STR独特的突变机制和更高的多态性所致。本研究为未来基于STR的牛基因组进化和选择研究奠定了基础。