Yan Haidan, Guan Qingzhou, He Jun, Lin Yunqing, Zhang Juan, Li Hongdong, Liu Huaping, Gu Yunyan, Guo Zheng, He Fei
Department of Systems Biology, College of Bioinformatics Science and Technology, Harbin Medical University, Harbin, 150086, China.
Key Laboratory of Ministry of Education for Gastrointestinal Cancer, Department of Bioinformatics, Fujian Medical University, Fuzhou, 350001, China.
J Transl Med. 2017 Feb 8;15(1):26. doi: 10.1186/s12967-017-1122-y.
Due to the heterogeneity of cancer, identifying differentially methylated (DM) CpG sites between a set of cancer samples and a set of normal samples cannot tell us which patients have methylation aberrations in a particular DM CpG site.
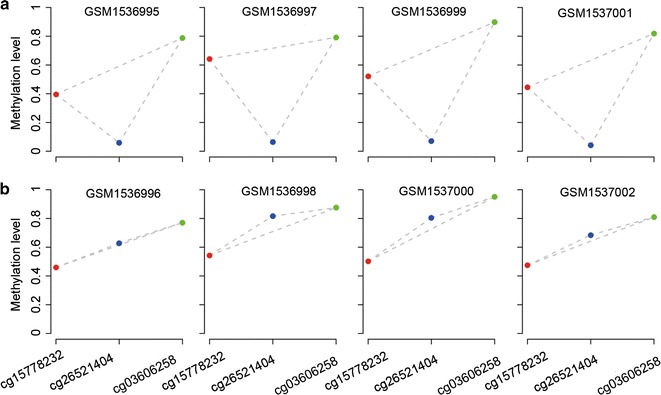
We firstly showed that the relative methylation-level orderings (RMOs) of CpG sites within individual normal lung tissues are highly stable but widely disrupted in lung adenocarcinoma tissues. This finding provides the basis of using the RankComp algorithm, previously developed for differential gene expression analysis at the individual level, to identify DM CpG sites in each cancer tissue compared with its own normal state. Briefly, through comparing with the highly stable normal RMOs predetermined in a large collection of samples for normal lung tissues, the algorithm finds those CpG sites whose hyper- or hypo-methylations may lead to the disrupted RMOs of CpG site pairs within a disease sample based on Fisher's exact test.
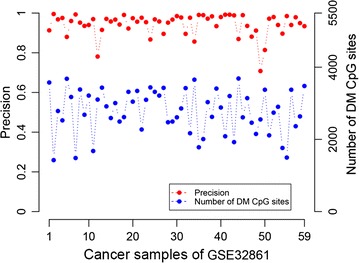
Evaluated in 59 lung adenocarcinoma tissues with paired adjacent normal tissues, RankComp reached an average precision of 94.26% for individual-level DM CpG sites. Then, after identifying DM CpG sites in each of the 539 lung adenocarcinoma samples from TCGA, we found five and 44 CpG sites hypermethylated and hypomethylated in above 90% of the disease samples, respectively. These findings were validated in 140 publicly available and eight additionally measured paired cancer-normal samples. Gene expression analysis revealed that four of the five genes, HOXA9, TAL1, ATP8A2, ENG and SPARCL1, each harboring one of the five frequently hypermethylated CpG sites within its promoters, were also frequently down-regulated in lung adenocarcinoma.
The common DNA methylation aberrations in lung adenocarcinoma tissues may be important for lung adenocarcinoma diagnosis and therapy.
由于癌症的异质性,在一组癌症样本和一组正常样本之间鉴定差异甲基化(DM)CpG位点并不能告诉我们哪些患者在特定的DM CpG位点存在甲基化异常。
我们首先表明,个体正常肺组织中CpG位点的相对甲基化水平排序(RMO)高度稳定,但在肺腺癌组织中广泛紊乱。这一发现为使用先前开发用于个体水平差异基因表达分析的RankComp算法奠定了基础,该算法用于识别每个癌症组织与其自身正常状态相比的DM CpG位点。简而言之,通过与在大量正常肺组织样本中预先确定的高度稳定的正常RMO进行比较,该算法基于Fisher精确检验找到那些高甲基化或低甲基化可能导致疾病样本中CpG位点对的RMO紊乱的CpG位点。
在59例伴有配对相邻正常组织的肺腺癌组织中进行评估,RankComp对个体水平的DM CpG位点的平均精度达到94.26%。然后,在鉴定了来自TCGA的539例肺腺癌样本中每一个的DM CpG位点后,我们发现分别有5个和44个CpG位点在超过90%的疾病样本中发生高甲基化和低甲基化。这些发现在140个公开可用的以及另外测量的8对癌症-正常样本中得到验证。基因表达分析显示,五个基因HOXA9、TAL1、ATP8A2、ENG和SPARCL1中的四个,其启动子内含有五个频繁高甲基化的CpG位点之一,在肺腺癌中也经常下调。
肺腺癌组织中常见的DNA甲基化异常可能对肺腺癌的诊断和治疗具有重要意义。