Wu Changchun, Xie Xueqin, Yang Xin, Du Mengze, Lin Hao, Huang Jian
The Clinical Hospital of Chengdu Brain Science Institute, School of Life Science and Technology, University of Electronic Science and Technology of China, Chengdu, 611731, China.
School of Healthcare Technology, Chengdu Neusoft University, Chengdu, 611844, China.
Mol Biomed. 2025 Apr 9;6(1):22. doi: 10.1186/s43556-025-00263-w.
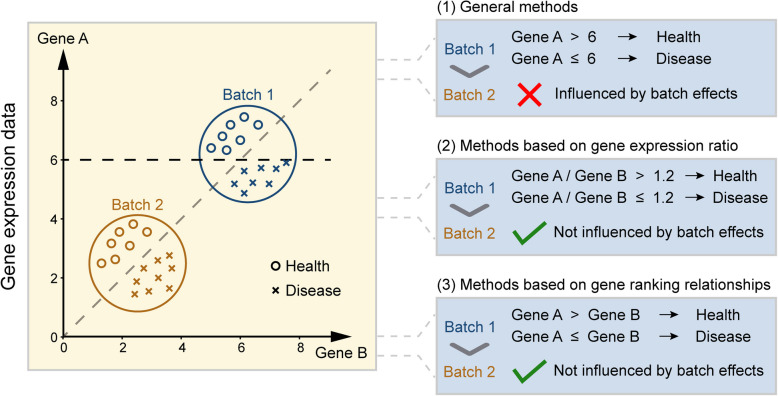
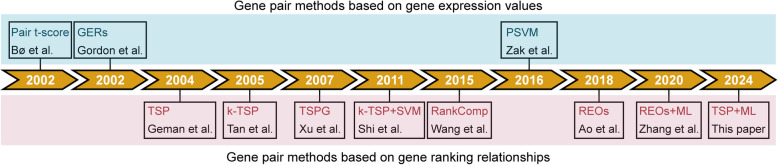
The rapid evolution of high-throughput sequencing technologies has revolutionized biomedical research, producing vast amounts of gene expression data that hold immense potential for biological discovery and clinical applications. Effectively mining these large-scale, high-dimensional data is crucial for facilitating disease detection, subtype differentiation, and understanding the molecular mechanisms underlying disease progression. However, the conventional paradigm of single-gene profiling, measuring absolute expression levels of individual genes, faces critical limitations in clinical implementation. These include vulnerability to batch effects and platform-dependent normalization requirements. In contrast, emerging approaches analyzing relative expression relationships between gene pairs demonstrate unique advantages. By focusing on binary comparisons of two genes' expression magnitudes, these methods inherently normalize experimental variations while capturing biologically stable interaction patterns. In this review, we systematically evaluate gene pair-based analytical frameworks. We classify eleven computational approaches into two fundamental categories: expression value-based methods quantifying differential expression patterns, and rank-based methods exploiting transcriptional ordering relationships. To bridge methodological development with practical implementation, we establish a reproducible analytical pipeline incorporating feature selection, classifier construction, and model evaluation modules using real-world benchmark datasets from pulmonary tuberculosis studies. These findings position gene pair analysis as a transformative paradigm for mining high-dimensional omics data, with direct implications for precision biomarker discovery and mechanistic studies of disease progression.
高通量测序技术的快速发展彻底改变了生物医学研究,产生了大量的基因表达数据,这些数据在生物学发现和临床应用方面具有巨大潜力。有效挖掘这些大规模、高维数据对于促进疾病检测、亚型区分以及理解疾病进展的分子机制至关重要。然而,测量单个基因绝对表达水平的单基因分析传统模式在临床应用中面临关键限制。这些限制包括易受批次效应影响以及依赖平台的标准化要求。相比之下,分析基因对之间相对表达关系的新兴方法显示出独特优势。通过关注两个基因表达量的二元比较,这些方法在捕获生物学稳定相互作用模式的同时,内在地对实验变异进行了标准化。在本综述中,我们系统地评估了基于基因对的分析框架。我们将11种计算方法分为两个基本类别:基于表达值的方法用于量化差异表达模式,以及基于排序的方法用于利用转录顺序关系。为了将方法学发展与实际应用联系起来,我们使用来自肺结核研究的真实基准数据集,建立了一个包含特征选择、分类器构建和模型评估模块的可重复分析流程。这些发现将基因对分析定位为挖掘高维组学数据的变革性模式,对精准生物标志物发现和疾病进展机制研究具有直接影响。