Kugelman Jeffrey R, Wiley Michael R, Nagle Elyse R, Reyes Daniel, Pfeffer Brad P, Kuhn Jens H, Sanchez-Lockhart Mariano, Palacios Gustavo F
Center for Genome Sciences, United States Army Medical Research Institute of Infectious Diseases (USAMRIID), Fort Detrick, Frederick, Maryland, United States of America.
Integrated Research Facility at Fort Detrick (IRF-Frederick), National Institute of Allergy and Infectious Diseases, National Institutes of Health, Fort Detrick, Frederick, Maryland, United States of America.
PLoS One. 2017 Feb 9;12(2):e0171333. doi: 10.1371/journal.pone.0171333. eCollection 2017.
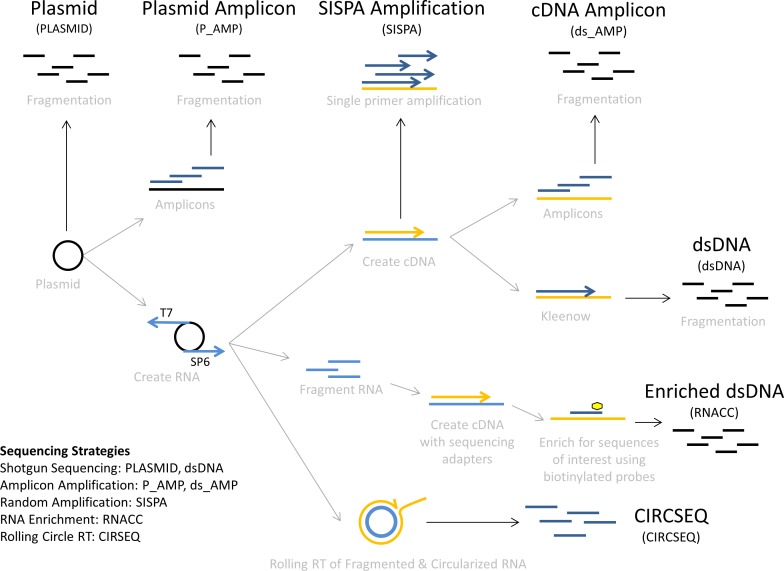
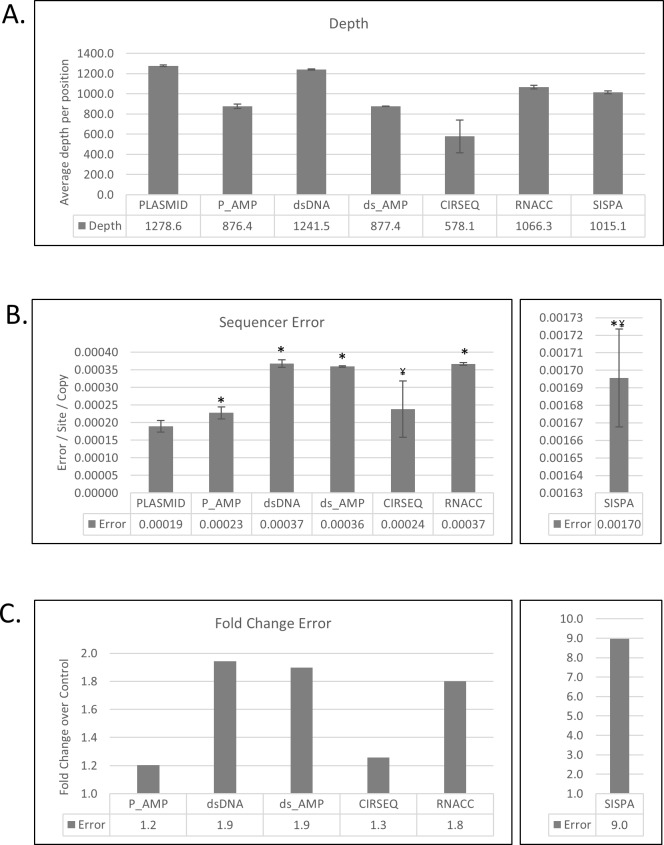
Individual RNA viruses typically occur as populations of genomes that differ slightly from each other due to mutations introduced by the error-prone viral polymerase. Understanding the variability of RNA virus genome populations is critical for understanding virus evolution because individual mutant genomes may gain evolutionary selective advantages and give rise to dominant subpopulations, possibly even leading to the emergence of viruses resistant to medical countermeasures. Reverse transcription of virus genome populations followed by next-generation sequencing is the only available method to characterize variation for RNA viruses. However, both steps may lead to the introduction of artificial mutations, thereby skewing the data. To better understand how such errors are introduced during sample preparation, we determined and compared error baseline rates of five different sample preparation methods by analyzing in vitro transcribed Ebola virus RNA from an artificial plasmid-based system. These methods included: shotgun sequencing from plasmid DNA or in vitro transcribed RNA as a basic "no amplification" method, amplicon sequencing from the plasmid DNA or in vitro transcribed RNA as a "targeted" amplification method, sequence-independent single-primer amplification (SISPA) as a "random" amplification method, rolling circle reverse transcription sequencing (CirSeq) as an advanced "no amplification" method, and Illumina TruSeq RNA Access as a "targeted" enrichment method. The measured error frequencies indicate that RNA Access offers the best tradeoff between sensitivity and sample preparation error (1.4-5) of all compared methods.
由于易出错的病毒聚合酶引入的突变,单个RNA病毒通常以基因组群体的形式出现,这些基因组群体彼此之间略有差异。了解RNA病毒基因组群体的变异性对于理解病毒进化至关重要,因为单个突变基因组可能获得进化选择优势并产生优势亚群体,甚至可能导致对医学对策产生抗性的病毒出现。对病毒基因组群体进行逆转录,然后进行下一代测序,是表征RNA病毒变异的唯一可用方法。然而,这两个步骤都可能导致引入人工突变,从而使数据产生偏差。为了更好地了解在样品制备过程中如何引入此类错误,我们通过分析基于人工质粒系统的体外转录埃博拉病毒RNA,确定并比较了五种不同样品制备方法的错误基线率。这些方法包括:从质粒DNA或体外转录RNA进行鸟枪法测序作为基本的“无扩增”方法,从质粒DNA或体外转录RNA进行扩增子测序作为“靶向”扩增方法,序列独立单引物扩增(SISPA)作为“随机”扩增方法,滚环逆转录测序(CirSeq)作为先进的“无扩增”方法,以及Illumina TruSeq RNA Access作为“靶向”富集方法。测量的错误频率表明,在所有比较的方法中,RNA Access在灵敏度和样品制备错误(1.4 - 5)之间提供了最佳平衡。