Ma Chunwei, Xu Shaohang, Liu Geng, Liu Xin, Xu Xun, Wen Bo, Liu Siqi
BGI-Shenzhen, Shenzhen, 518083, China.
BMC Bioinformatics. 2017 Feb 16;18(1):109. doi: 10.1186/s12859-017-1491-5.
Tandem mass spectrometry (MS/MS) followed by database search is a main approach to identify peptides/proteins in proteomic studies. A lot of effort has been devoted to improve the identification accuracy and sensitivity for peptides/proteins, such as developing advanced algorithms and expanding protein databases.
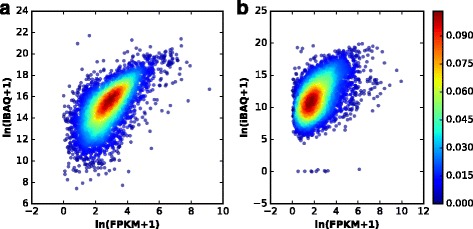
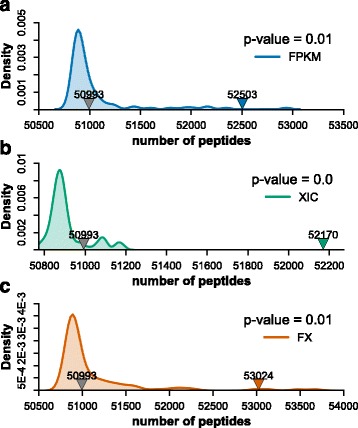
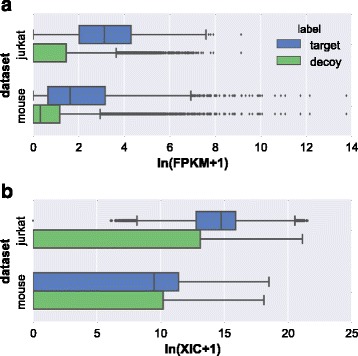
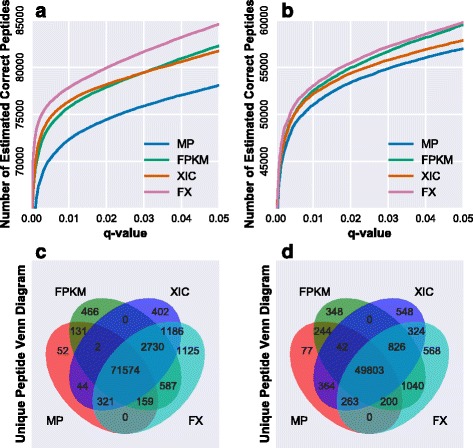
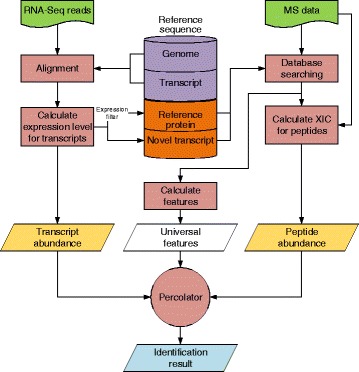
Herein, we described a new strategy for enhancing the sensitivity of protein/peptide identification through combination of mRNA and peptide abundance in Percolator. In our strategy, a new workflow for peptide identification is established on the basis of the abundance of transcripts and potential novel transcripts derived from RNA-Seq and abundance of peptides towards the same life species. We demonstrate the utility of this strategy by two MS/MS datasets and the results indicate that about 5% ~ 8% improvement of peptide identification can be achieved with 1% FDR in peptide level by integrating the peptide abundance, the transcript abundance and potential novel transcripts from RNA-Seq data. Meanwhile, 181 and 154 novel peptides were identified in the two datasets, respectively.
We have demonstrated that this strategy could enable improvement of peptide/protein identification and discovery of novel peptides, as compared with the traditional search methods.
串联质谱(MS/MS)结合数据库搜索是蛋白质组学研究中鉴定肽段/蛋白质的主要方法。人们已经付出了很多努力来提高肽段/蛋白质的鉴定准确性和灵敏度,例如开发先进的算法和扩展蛋白质数据库。
在此,我们描述了一种通过结合Percolator中mRNA和肽段丰度来提高蛋白质/肽段鉴定灵敏度的新策略。在我们的策略中,基于来自RNA测序的转录本丰度和潜在新转录本以及同一生物物种的肽段丰度,建立了一种新的肽段鉴定工作流程。我们通过两个MS/MS数据集证明了该策略的实用性,结果表明,通过整合肽段丰度、转录本丰度和来自RNA测序数据的潜在新转录本,在肽段水平上以1%的错误发现率(FDR)可实现约5%至8%的肽段鉴定改进。同时,在这两个数据集中分别鉴定出181个和154个新肽段。
我们已经证明,与传统搜索方法相比,该策略能够改进肽段/蛋白质鉴定并发现新肽段。