Department of Genetics, Texas Biomedical Research Institute, San Antonio, Texas, USA.
Department of Chemistry, University of Wisconsin, Madison, Wisconsin, USA.
BMC Genomics. 2017 Nov 13;18(1):877. doi: 10.1186/s12864-017-4279-0.
Shotgun proteomics utilizes a database search strategy to compare detected mass spectra to a library of theoretical spectra derived from reference genome information. As such, the robustness of proteomics results is contingent upon the completeness and accuracy of the gene annotation in the reference genome. For animal models of disease where genomic annotation is incomplete, such as non-human primates, proteogenomic methods can improve the detection of proteins by incorporating transcriptional data from RNA-Seq to improve proteomics search databases used for peptide spectral matching. Customized search databases derived from RNA-Seq data are capable of identifying unannotated genetic and splice variants while simultaneously reducing the number of comparisons to only those transcripts actively expressed in the tissue.
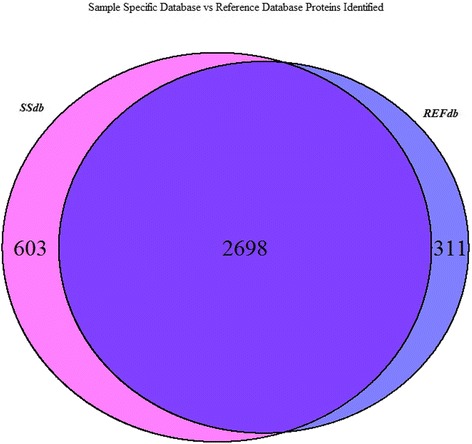
We collected RNA-Seq and proteomic data from 10 vervet monkey liver samples and used the RNA-Seq data to curate sample-specific search databases which were analyzed in the program Morpheus. We compared these results against those from a search database generated from the reference vervet genome. A total of 284 previously unannotated splice junctions were predicted by the RNA-Seq data, 92 of which were confirmed by peptide spectral matches. More than half (53/92) of these unannotated splice variants had orthologs in other non-human primates, suggesting that failure to match these peptides in the reference analyses likely arose from incomplete gene model information. The sample-specific databases also identified 101 unique peptides containing single amino acid substitutions which were missed by the reference database. Because the sample-specific searches were restricted to actively expressed transcripts, the search databases were smaller, more computationally efficient, and identified more peptides at the empirically derived 1 % false discovery rate.
Proteogenomic approaches are ideally suited to facilitate the discovery and annotation of proteins in less widely studies animal models such as non-human primates. We expect that these approaches will help to improve existing genome annotations of non-human primate species such as vervet.
shotgun 蛋白质组学利用数据库搜索策略将检测到的质谱与从参考基因组信息衍生的理论光谱库进行比较。因此,蛋白质组学结果的可靠性取决于参考基因组中基因注释的完整性和准确性。对于疾病的动物模型,如非人类灵长类动物,基因组注释不完整,蛋白质基因组学方法可以通过整合 RNA-Seq 的转录数据来改进蛋白质组学搜索数据库,从而提高肽谱匹配中蛋白质的检测。从 RNA-Seq 数据中衍生的定制搜索数据库能够识别未注释的遗传和剪接变体,同时将比较数量减少到仅针对组织中活跃表达的转录本。
我们从 10 只长尾猕猴肝脏样本中收集了 RNA-Seq 和蛋白质组学数据,并使用 RNA-Seq 数据来整理特定于样本的搜索数据库,这些数据库在程序 Morpheus 中进行了分析。我们将这些结果与从参考长尾猕猴基因组生成的搜索数据库进行了比较。RNA-Seq 数据共预测了 284 个先前未注释的剪接接头,其中 92 个被肽谱匹配证实。这些未注释的剪接变体中有一半以上(53/92)在其他非人类灵长类动物中有同源物,这表明在参考分析中未能匹配这些肽很可能是由于基因模型信息不完整。特定于样本的数据库还鉴定了 101 个含有单个氨基酸替换的独特肽,这些肽在参考数据库中被遗漏。由于特定于样本的搜索仅限于活跃表达的转录本,因此搜索数据库更小,计算效率更高,并且在经验上确定的 1%假发现率下鉴定了更多的肽。
蛋白质基因组学方法非常适合促进在研究较少的动物模型(如非人类灵长类动物)中发现和注释蛋白质。我们期望这些方法将有助于改进非人类灵长类物种(如长尾猕猴)的现有基因组注释。