Biomolecular Mass Spectrometry and Proteomics Group, Bijvoet Center for Biomolecular Research, Utrecht University , Padualaan 8, 3584 CH Utrecht, The Netherlands.
Netherlands Proteomics Center , Padualaan 8, 3584 CH Utrecht, The Netherlands.
Anal Chem. 2017 Mar 21;89(6):3318-3325. doi: 10.1021/acs.analchem.6b03756. Epub 2017 Mar 10.
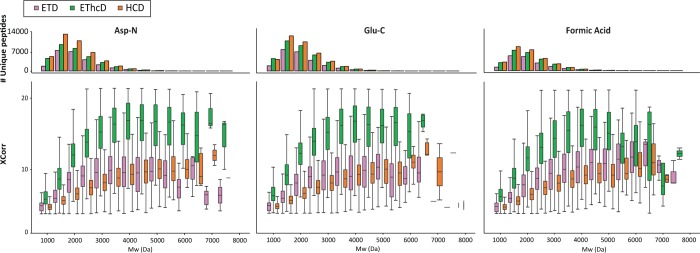
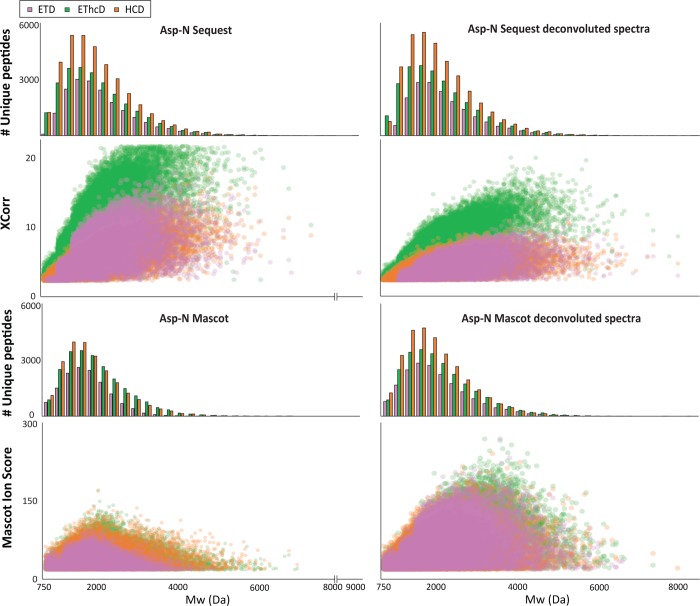
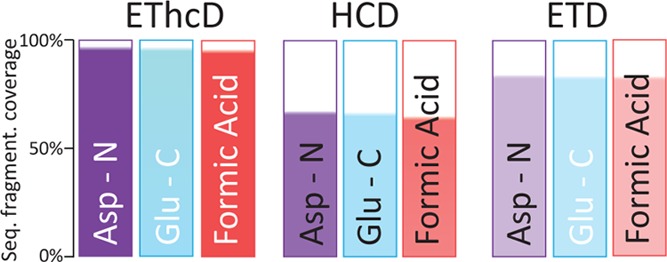
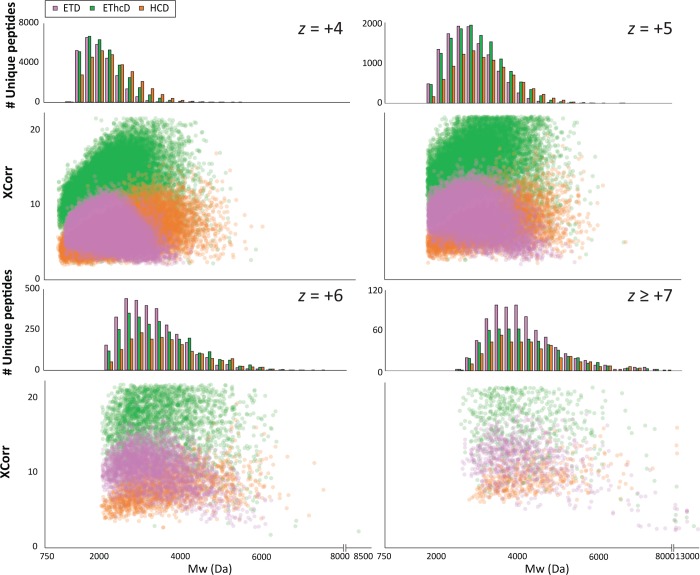
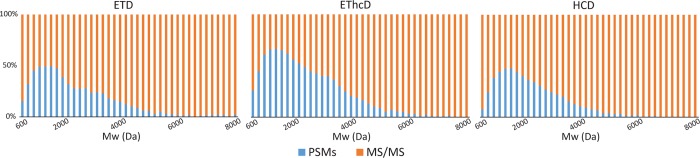
Mass spectrometry (MS)-based proteomics workflows can crudely be classified into two distinct regimes, targeting either relatively small peptides (i.e., 0.7 kDa < M < 3.0 kDa) or small to medium sized intact proteins (i.e., 10 kDa < M < 30 kDa), respectively, termed bottom-up and top-down proteomics. Recently, a niche has started to be explored covering the analysis of middle-range peptides (i.e., 3.0 kDa < M < 10 kDa), aptly termed middle-down proteomics. Although middle-down proteomics can follow, in principle, a modular workflow similar to that of bottom-up proteomics, we hypothesized that each of these modules would benefit from targeted optimization to improve its overall performance in the analysis of middle-range sized peptides. Hence, to generate middle-range sized peptides from cellular lysates, we explored the use of the proteases Asp-N and Glu-C and a nonenzymatic acid induced cleavage. To increase the depth of the proteome, a strong cation exchange (SCX) separation, carefully tuned to improve the separation of longer peptides, combined with reversed phase-liquid chromatography (RP-LC) using columns packed with material possessing a larger pore size, was used. Finally, after evaluating the combination of potentially beneficial MS settings, we also assessed the peptide fragmentation techniques, including higher-energy collision dissociation (HCD), electron-transfer dissociation (ETD), and electron-transfer combined with higher-energy collision dissociation (EThcD), for characterization of middle-range sized peptides. These combined improvements clearly improve the detection and sequence coverage of middle-range peptides and should guide researchers to explore further how middle-down proteomics may lead to an improved proteome coverage, beneficial for, among other things, the enhanced analysis of (co-occurring) post-translational modifications.
基于质谱(MS)的蛋白质组学工作流程可以粗略地分为两种截然不同的模式,分别针对相对较小的肽(即 0.7 kDa < M < 3.0 kDa)或小到中等大小的完整蛋白质(即 10 kDa < M < 30 kDa),分别称为自上而下和自下而上的蛋白质组学。最近,一个利基市场开始被探索,涵盖了中等范围肽(即 3.0 kDa < M < 10 kDa)的分析,恰当地称为中间向下蛋白质组学。虽然中间向下蛋白质组学在原则上可以遵循类似于自上而下蛋白质组学的模块化工作流程,但我们假设这些模块中的每一个都将受益于有针对性的优化,以提高其在分析中等范围大小肽中的整体性能。因此,为了从细胞裂解物中生成中等范围大小的肽,我们探索了使用 Asp-N 和 Glu-C 蛋白酶以及非酶促酸诱导切割。为了增加蛋白质组的深度,使用强阳离子交换(SCX)分离,仔细调整以改善较长肽的分离,与使用具有较大孔径的填充材料的反相液相色谱(RP-LC)相结合。最后,在评估了潜在有益的 MS 设置的组合之后,我们还评估了肽片段化技术,包括更高能量碰撞解离(HCD)、电子转移解离(ETD)和电子转移与更高能量碰撞解离的组合(EThcD),用于中间范围大小的肽的特征描述。这些综合改进明显提高了中等范围肽的检测和序列覆盖度,应该指导研究人员进一步探索中间向下蛋白质组学如何可能导致蛋白质组覆盖度的提高,这对于增强(共发生)翻译后修饰的分析等方面非常有益。