Maruyama Toru, Mori Tetsushi, Yamagishi Keisuke, Takeyama Haruko
Department of Life Science & Medical Bioscience, Graduate School of Advanced Science & Engineering, Waseda University, 3-4-1 Okubo, Shinjuku, Tokyo, 169-8555, Japan.
Computational Bio-Big Data Open Innovation Lab., National Institute of Advanced Science and Technology, 3-4-1 Okubo, Shinjuku, Tokyo, 169-0072, Japan.
BMC Bioinformatics. 2017 Mar 4;18(1):152. doi: 10.1186/s12859-017-1572-5.
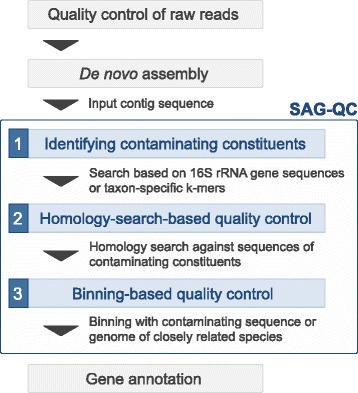
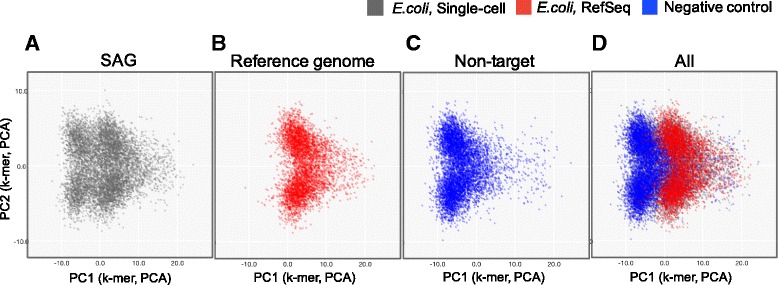
Whole genome amplification techniques have enabled the analysis of unexplored genomic information by sequencing of single-amplified genomes (SAGs). Whole genome amplification of single bacteria is currently challenging because contamination often occurs in experimental processes. Thus, to increase the confidence in the analyses of sequenced SAGs, bioinformatics approaches that identify and exclude non-target sequences from SAGs are required. Since currently reported approaches utilize sequence information in public databases, they have limitations when new strains are the targets of interest. Here, we developed a software SAG-QC that identify and exclude non-target sequences independent of database.
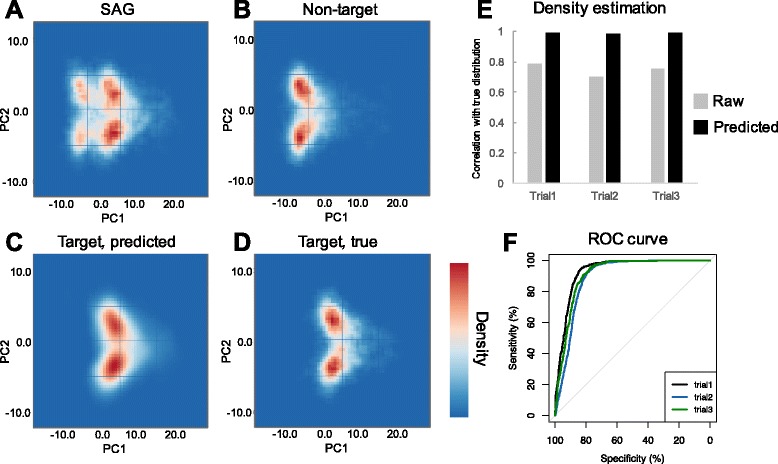
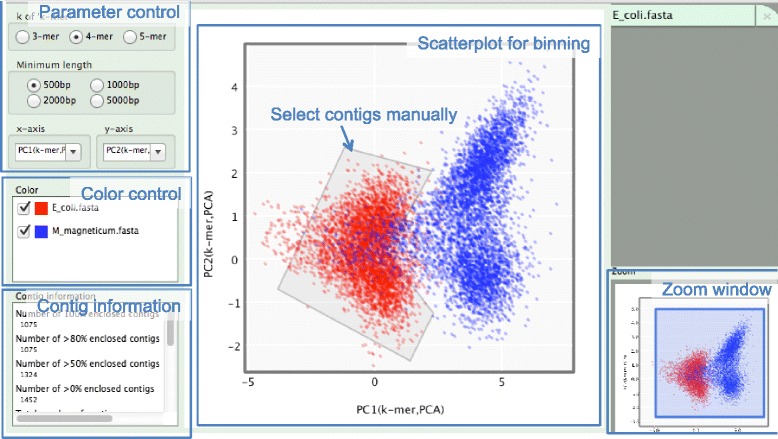
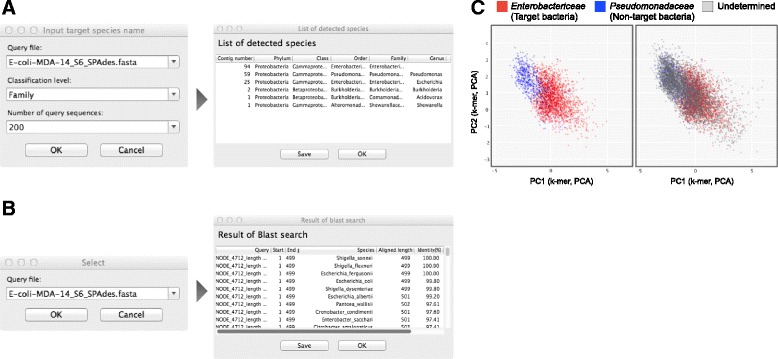
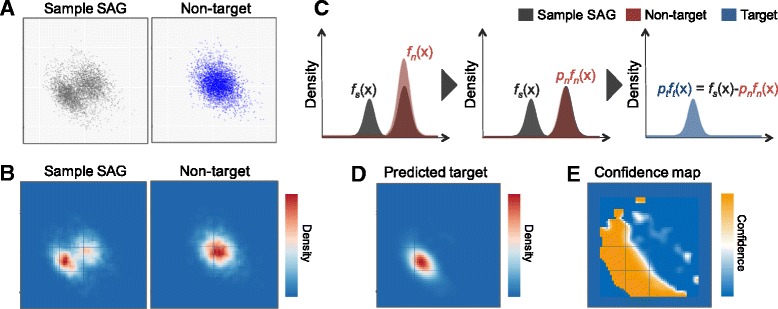
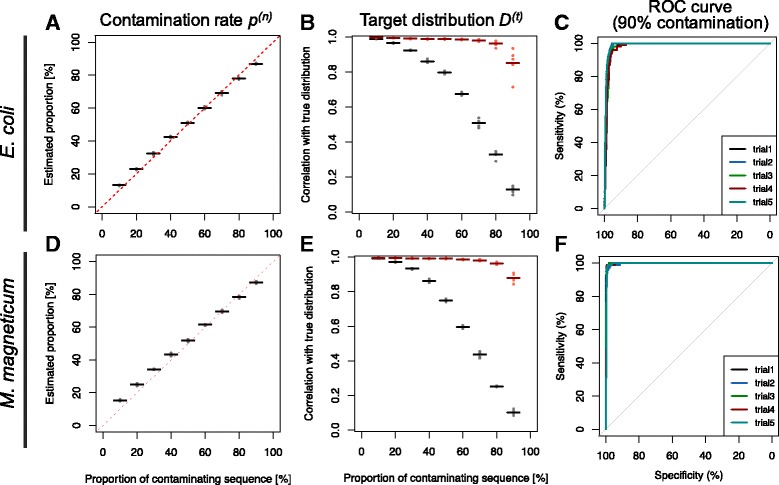
In our method, "no template control" sequences acquired during WGA were used. We calculated the probability that a sequence was derived from contaminants by comparing k-mer compositions with the no template control sequences. Based on the results of tests using simulated SAG datasets, the accuracy of our method for predicting non-target sequences was higher than that of currently reported techniques. Subsequently, we applied our tool to actual SAG datasets and evaluated the accuracy of the predictions.
Our method works independently of public sequence information for distinguishing SAGs from non-target sequences. This method will be effective when employed against SAG sequences of unexplored strains and we anticipate that it will contribute to the correct interpretation of SAGs.
全基因组扩增技术使得通过对单扩增基因组(SAGs)进行测序来分析未探索的基因组信息成为可能。目前,对单个细菌进行全基因组扩增具有挑战性,因为在实验过程中经常会发生污染。因此,为了提高对测序SAGs分析结果的可信度,需要采用生物信息学方法来识别并排除SAGs中的非目标序列。由于目前报道的方法利用公共数据库中的序列信息,当新菌株是感兴趣的目标时,它们存在局限性。在此,我们开发了一款名为SAG-QC的软件,该软件能够独立于数据库识别并排除非目标序列。
在我们的方法中,使用了全基因组扩增过程中获得的“无模板对照”序列。通过将k-mer组成与无模板对照序列进行比较,我们计算了一个序列源自污染物的概率。基于使用模拟SAG数据集进行测试的结果,我们的方法预测非目标序列的准确性高于目前报道的技术。随后,我们将我们的工具应用于实际的SAG数据集,并评估了预测的准确性。
我们的方法独立于公共序列信息来区分SAGs和非目标序列。当应用于未探索菌株的SAG序列时,该方法将是有效的,并且我们预计它将有助于对SAGs进行正确的解读。