Cifani Paolo, Shakiba Mojdeh, Chhangawala Sagar, Kentsis Alex
Molecular Pharmacology Program, Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center, New York, NY, USA.
Physiology, Biophysics and Systems Biology Program, Weill Cornell Graduate School of Medical Sciences, New York, NY, USA.
BMC Bioinformatics. 2017 Mar 4;18(1):153. doi: 10.1186/s12859-017-1563-6.
High-accuracy mass spectrometry enables near comprehensive quantification of the components of the cellular proteomes, increasingly including their chemically modified variants. Likewise, large-scale libraries of quantified synthetic peptides are becoming available, enabling absolute quantification of chemically modified proteoforms, and therefore systems-level analyses of changes of their absolute abundance and stoichiometry. Existing computational methods provide advanced tools for mass spectral analysis and statistical inference, but lack integrated functions for quantitative analysis of post-translationally modified proteins and their modification stoichiometry.
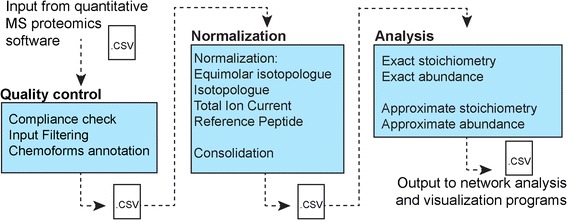
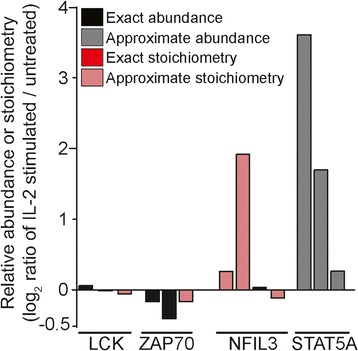
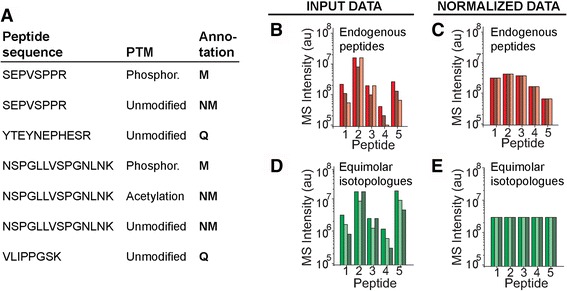
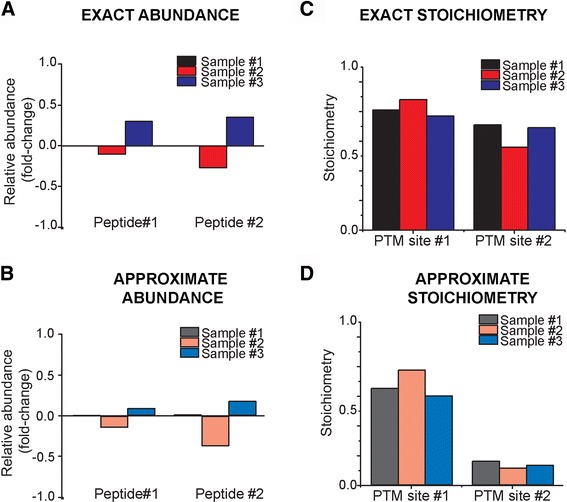
Here, we develop ProteoModlR, a program for quantitative analysis of abundance and stoichiometry of post-translational chemical modifications across temporal and steady-state biological states. While ProteoModlR is intended for the analysis of experiments using isotopically labeled reference peptides for absolute quantitation, it also supports the analysis of labeled and label-free data, acquired in both data-dependent and data-independent modes for relative quantitation. Moreover, ProteoModlR enables functional analysis of sparsely sampled quantitative mass spectrometry experiments by inferring the missing values from the available measurements, without imputation. The implemented architecture includes parsing and normalization functions to control for common sources of technical variation. Finally, ProteoModlR's modular design and interchangeable format are optimally suited for integration with existing computational proteomics tools, thereby facilitating comprehensive quantitative analysis of cellular signaling.
ProteoModlR and its documentation are available for download at http://github.com/kentsisresearchgroup/ProteoModlR as a stand-alone R package.
高精度质谱能够近乎全面地定量细胞蛋白质组的成分,越来越多地包括其化学修饰变体。同样,大规模的定量合成肽库也已可用,这使得对化学修饰蛋白质形式进行绝对定量成为可能,从而能够对其绝对丰度和化学计量的变化进行系统水平的分析。现有的计算方法为质谱分析和统计推断提供了先进工具,但缺乏对翻译后修饰蛋白质及其修饰化学计量进行定量分析的集成功能。
在此,我们开发了ProteoModlR,这是一个用于定量分析跨时间和稳态生物状态的翻译后化学修饰的丰度和化学计量的程序。虽然ProteoModlR旨在分析使用同位素标记参考肽进行绝对定量的实验,但它也支持对以数据依赖和数据独立模式获取的标记和无标记数据进行分析,以进行相对定量。此外,ProteoModlR能够通过从可用测量值推断缺失值来对稀疏采样的定量质谱实验进行功能分析,而无需插补。所实现的架构包括解析和归一化功能,以控制技术变异的常见来源。最后,ProteoModlR的模块化设计和可互换格式最适合与现有的计算蛋白质组学工具集成,从而便于对细胞信号传导进行全面的定量分析。
ProteoModlR及其文档可在http://github.com/kentsisresearchgroup/ProteoModlR上作为独立的R包下载。