Kalvet Indrek, Guo Qianqian, Tizzard Graham J, Schoenebeck Franziska
Institute of Organic Chemistry, RWTH Aachen University , Landoltweg 1, 52074 Aachen, Germany.
Institute of Inorganic Chemistry, X-ray Crystallography, RWTH Aachen University , Landoltweg 1, 52074 Aachen, Germany.
ACS Catal. 2017 Mar 3;7(3):2126-2132. doi: 10.1021/acscatal.6b03344. Epub 2017 Jan 31.
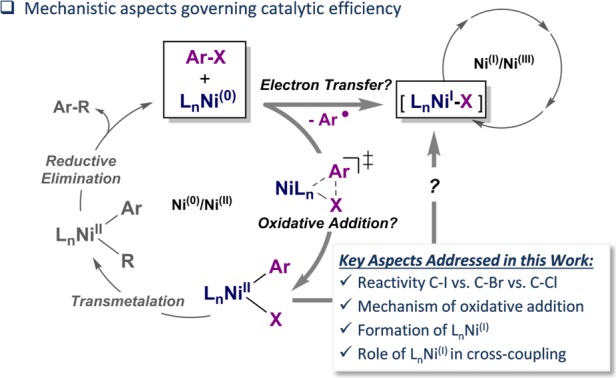
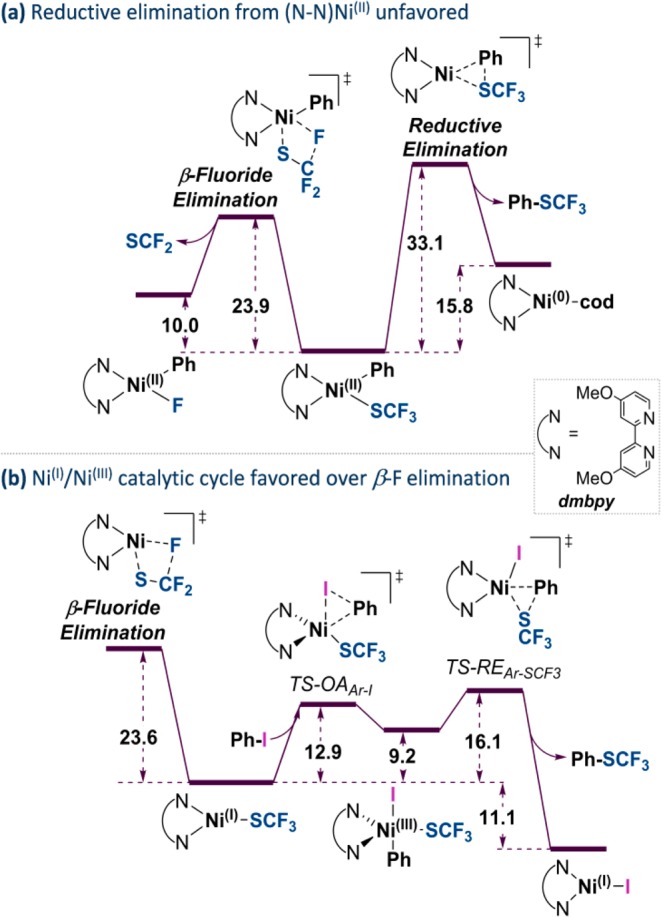
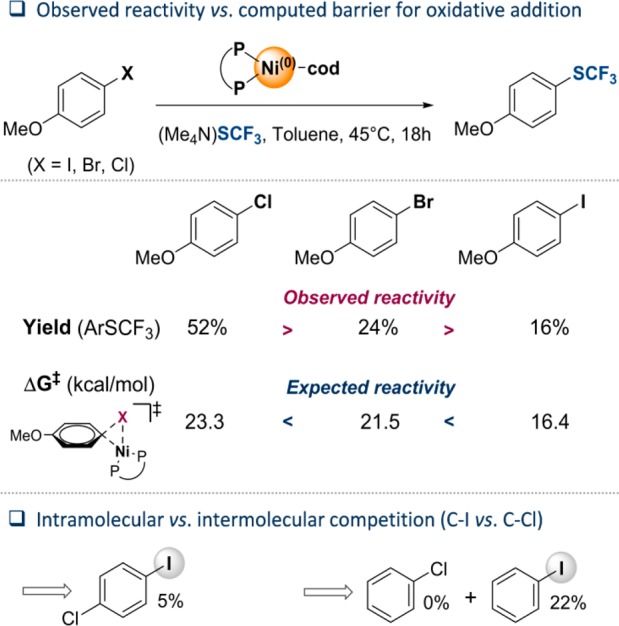
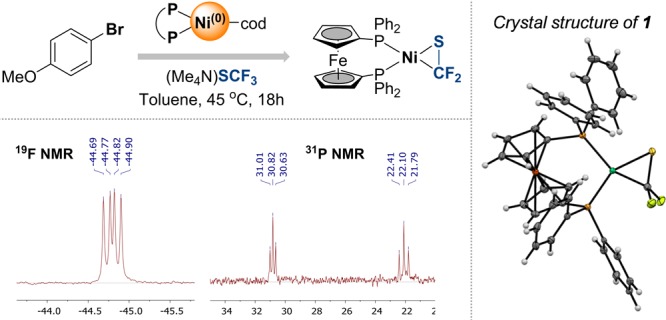
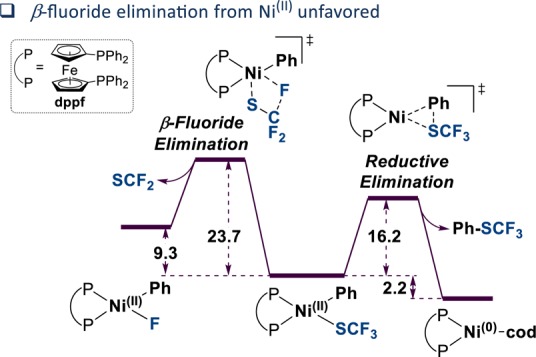
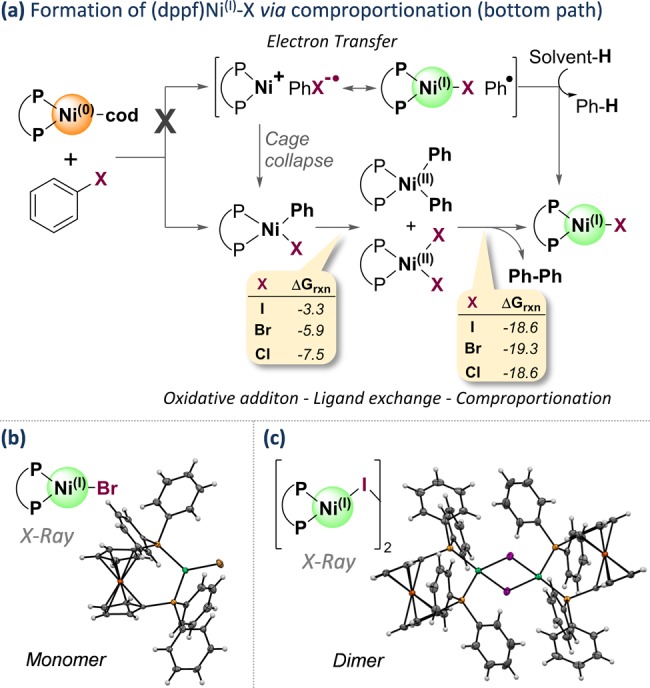
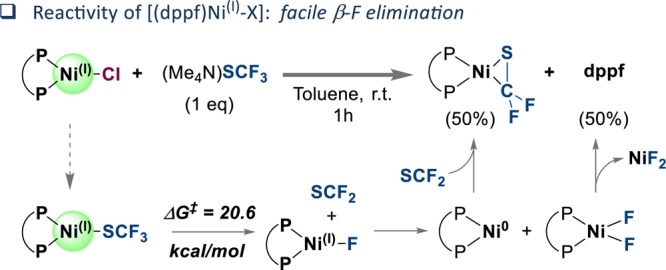
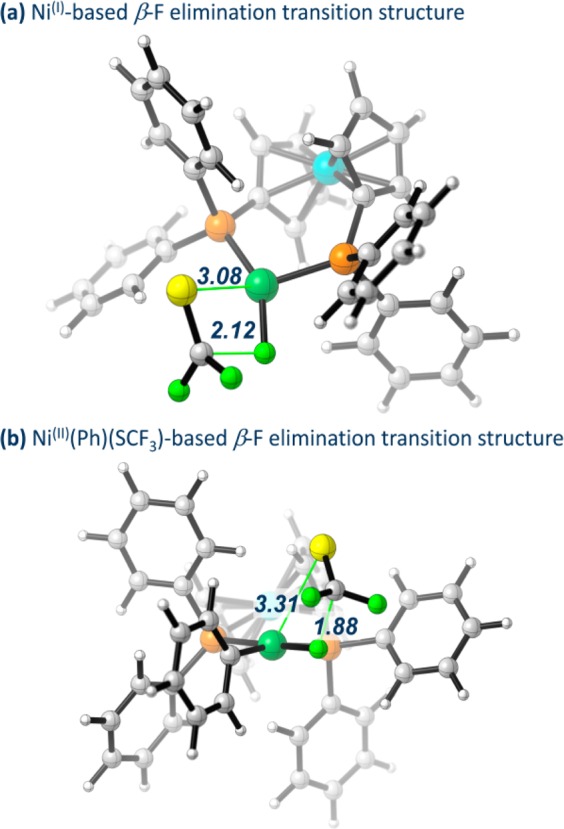
The direct introduction of the valuable SCF moiety into organic molecules has received considerable attention. While it can be achieved successfully for aryl chlorides under catalysis with Ni(cod) and dppf, this report investigates the Ni-catalyzed functionalization of the seemingly more reactive aryl halides ArI and ArBr. Counterintuitively, the observed conversion triggered by dppf/Ni is ArCl > ArBr > ArI, at odds with bond strength preferences. By a combined computational and experimental approach, the origin of this was identified to be due to the formation of (dppf)Ni, which favors β-F elimination as a competing pathway over the productive cross-coupling, ultimately generating the inactive complex (dppf)Ni(SCF) as a catalysis dead end. The complexes (dppf)Ni-Br and (dppf)Ni-I were isolated and resolved by X-ray crystallography. Their formation was found to be consistent with a ligand-exchange-induced comproportionation mechanism. In stark contrast to these phosphine-derived Ni complexes, the corresponding nitrogen-ligand-derived species were found to be likely competent catalysts in oxidation state I. Our computational studies of N-ligand derived Ni complexes fully support productive Ni/Ni catalysis, as the competing β-F elimination is disfavored. Moreover, N-derived Ni complexes are predicted to be more reactive than their Ni counterparts in catalysis. These data showcase fundamentally different roles of Ni in carbon-heteroatom bond formation depending on the ligand sphere.
将有价值的SCF部分直接引入有机分子已受到广泛关注。虽然在Ni(cod)和dppf催化下芳基氯可以成功实现这一过程,但本报告研究了看似更具反应性的芳基卤化物ArI和ArBr的镍催化官能化反应。与直觉相反,观察到由dppf/Ni引发的转化率为ArCl>ArBr>ArI,这与键强度偏好不一致。通过计算和实验相结合的方法,确定其原因是(dppf)Ni的形成,它有利于β-F消除作为一种竞争途径,而不是进行有效的交叉偶联,最终生成无活性的配合物(dppf)Ni(SCF)作为催化死端。通过X射线晶体学分离并解析了配合物(dppf)Ni-Br和(dppf)Ni-I。发现它们的形成与配体交换诱导的歧化机制一致。与这些膦衍生的镍配合物形成鲜明对比的是,相应的氮配体衍生的物种在氧化态I下可能是有效的催化剂。我们对氮配体衍生的镍配合物的计算研究完全支持有效的Ni/Ni催化,因为竞争的β-F消除不受青睐。此外,预测氮衍生的镍配合物在催化中比其镍对应物更具反应性。这些数据展示了镍在碳-杂原子键形成中根据配体环境所起的根本不同的作用。