Université Clermont Auvergne, INRA, MEDIS, 63000, Clermont-Ferrand, France.
Microbiome. 2017 Mar 14;5(1):33. doi: 10.1186/s40168-017-0251-0.
High-throughput DNA sequencing technologies have revolutionized genomic analysis, including the de novo assembly of whole genomes from single organisms or metagenomic samples. However, due to the limited capacity of short-read sequence data to assemble complex or low coverage regions, genomes are typically fragmented, leading to draft genomes with numerous underexplored large genomic regions. Revealing these missing sequences is a major goal to resolve concerns in numerous biological studies.
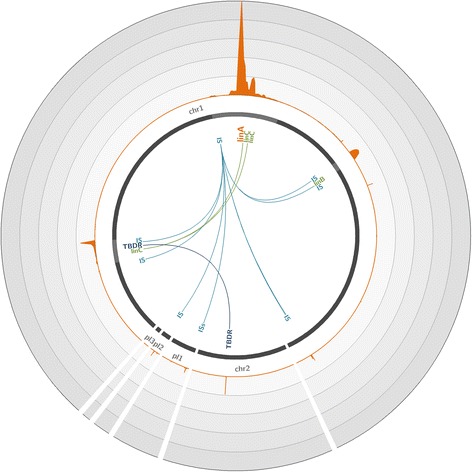
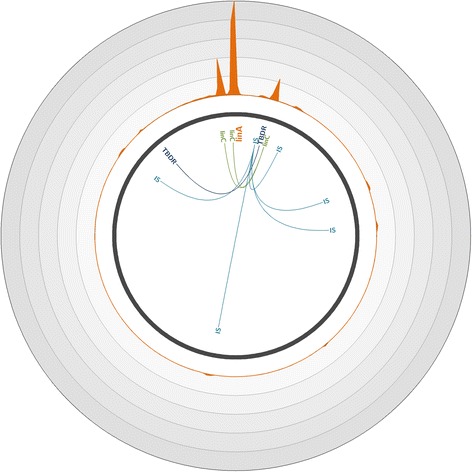
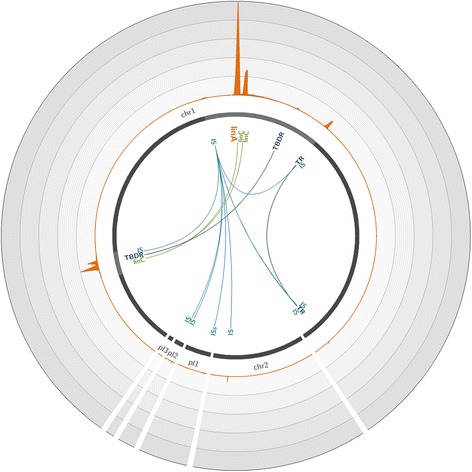
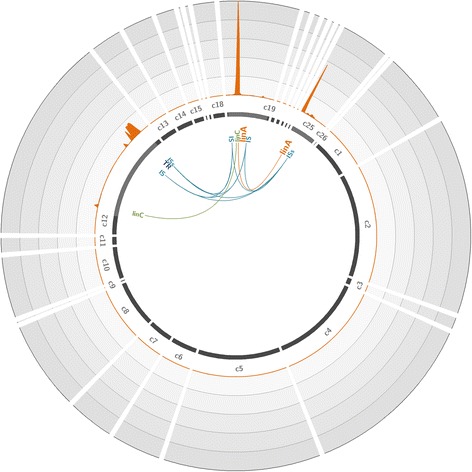
To overcome these limitations, we developed an innovative target enrichment method for the reconstruction of large unknown genomic regions. Based on a hybridization capture strategy, this approach enables the enrichment of large genomic regions allowing the reconstruction of tens of kilobase pairs flanking a short, targeted DNA sequence.
Applied to a metagenomic soil sample targeting the linA gene, the biomarker of hexachlorocyclohexane (HCH) degradation, our method permitted the enrichment of the gene and its flanking regions leading to the reconstruction of several contigs and complete plasmids exceeding tens of kilobase pairs surrounding linA. Thus, through gene association and genome reconstruction, we identified microbial species involved in HCH degradation which constitute targets to improve biostimulation treatments.
This new hybridization capture strategy makes surveying and deconvoluting complex genomic regions possible through large genomic regions enrichment and allows the efficient exploration of metagenomic diversity. Indeed, this approach enables to assign identity and function to microorganisms in natural environments, one of the ultimate goals of microbial ecology.
高通量 DNA 测序技术彻底改变了基因组分析,包括从单个生物或宏基因组样本中从头组装整个基因组。然而,由于短读序列数据在组装复杂或低覆盖区域方面的能力有限,基因组通常会被分割,导致具有许多未充分探索的大型基因组区域的草图基因组。揭示这些缺失的序列是解决众多生物学研究中关注问题的主要目标。
为了克服这些限制,我们开发了一种用于重建大型未知基因组区域的创新靶向富集方法。该方法基于杂交捕获策略,可富集大型基因组区域,从而重建数十个千碱基对侧翼的短靶向 DNA 序列。
将该方法应用于针对 linA 基因(六氯环己烷(HCH)降解的生物标志物)的宏基因组土壤样本,我们的方法实现了基因及其侧翼区域的富集,从而重建了数十个千碱基对以上的多个 contigs 和完整质粒,这些质粒围绕着 linA。因此,通过基因关联和基因组重建,我们鉴定出了参与 HCH 降解的微生物物种,它们是提高生物刺激处理的目标。
这种新的杂交捕获策略通过大型基因组区域的富集,使对复杂基因组区域的调查和解析成为可能,并允许对宏基因组多样性进行高效探索。事实上,这种方法能够对自然环境中的微生物进行身份和功能的鉴定,这是微生物生态学的最终目标之一。