Belsom Adam, Schneider Michael, Fischer Lutz, Mabrouk Mahmoud, Stahl Kolja, Brock Oliver, Rappsilber Juri
Wellcome Trust Centre for Cell Biology, University of Edinburgh, Edinburgh, EH9 3BF, UK.
Robotics and Biology Laboratory, Technische Universität Berlin, Berlin, 10587, Germany.
Wellcome Open Res. 2016 Dec 9;1:24. doi: 10.12688/wellcomeopenres.10046.1.
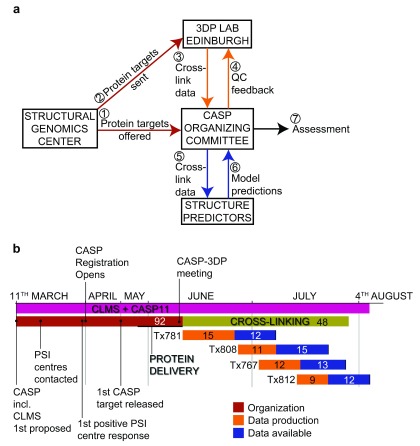
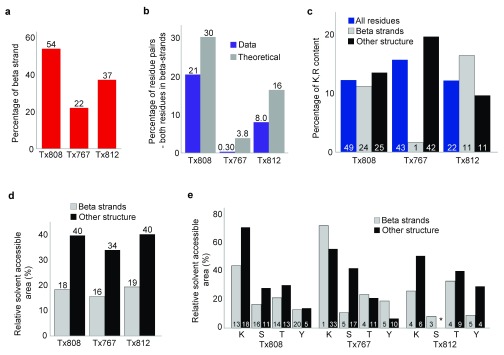
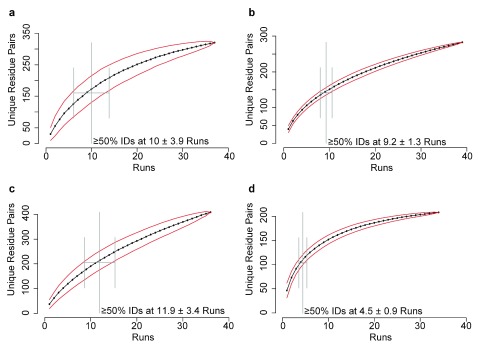
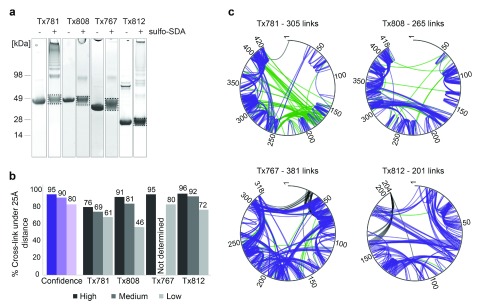
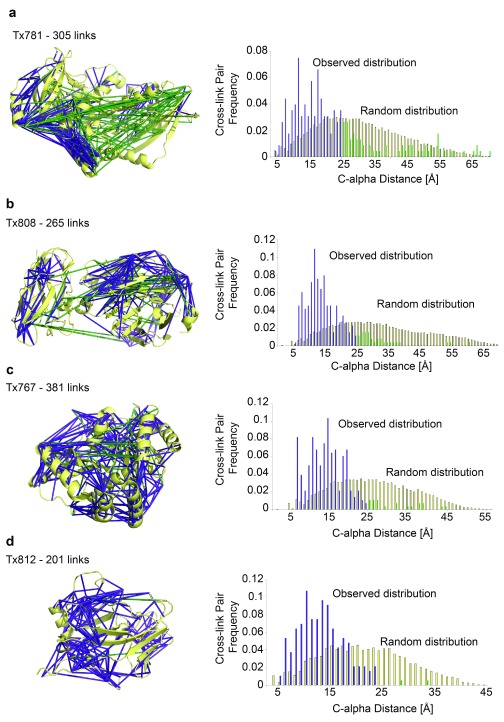
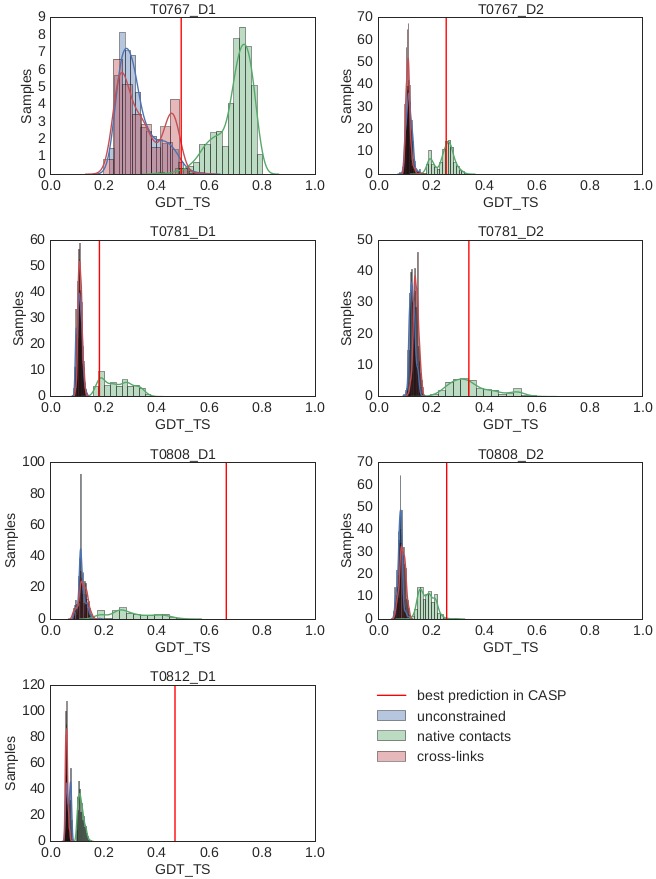
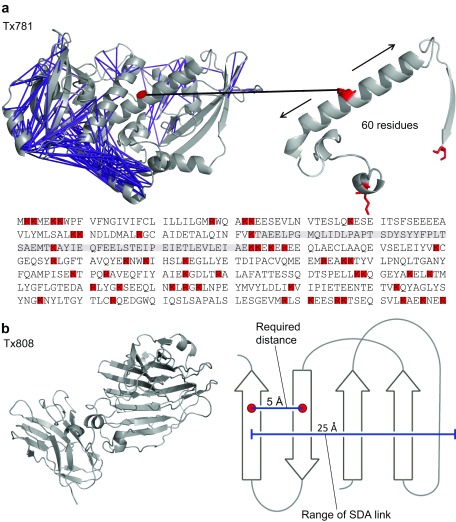
Determining the structure of a protein by any method requires various contributions from experimental and computational sides. In a recent study, high-density cross-linking/mass spectrometry (HD-CLMS) data in combination with structure prediction determined the structure of human serum albumin (HSA) domains, with an RMSD to X-ray structure of up to 2.5 Å, or 3.4 Å in the context of blood serum. This paper reports the blind test on the readiness of this technology through the help of Critical Assessment of protein Structure Prediction (CASP). We identified between 201-381 unique residue pairs at an estimated 5% FDR (at link level albeit with missing site assignment precision evaluation), for four target proteins. HD-CLMS proved reliable once crystal structures were released. However, improvements in structure prediction using cross-link data were slight. We identified two reasons for this. Spread of cross-links along the protein sequence and the tightness of the spatial constraints must be improved. However, for the selected targets even ideal contact data derived from crystal structures did not allow modellers to arrive at the observed structure. Consequently, the progress of HD-CLMS in conjunction with computational modeling methods as a structure determination method, depends on advances on both arms of this hybrid approach.
通过任何方法确定蛋白质的结构都需要实验和计算方面的各种贡献。在最近的一项研究中,高密度交联/质谱(HD-CLMS)数据与结构预测相结合,确定了人血清白蛋白(HSA)结构域的结构,与X射线结构的均方根偏差(RMSD)高达2.5 Å,在血清环境中为3.4 Å。本文借助蛋白质结构预测关键评估(CASP)报告了对该技术成熟度的盲测。对于四种目标蛋白质,我们在估计5%的错误发现率(FDR)下(在连接水平上,尽管缺少位点分配精度评估)鉴定出201-381个独特的残基对。一旦晶体结构公布,HD-CLMS被证明是可靠的。然而,使用交联数据进行结构预测的改进很小。我们确定了两个原因。交联在蛋白质序列上的分布以及空间约束的紧密性必须得到改善。然而,对于选定的目标,即使是从晶体结构获得的理想接触数据也不能让建模者得到观察到的结构。因此,HD-CLMS与计算建模方法相结合作为一种结构测定方法的进展,取决于这种混合方法两个方面的进展。