McTavish Emily Jane, Pettengill James, Davis Steven, Rand Hugh, Strain Errol, Allard Marc, Timme Ruth E
University of California, Merced, Merced, CA, USA.
University of Kansas, Lawrence, RS, USA.
BMC Bioinformatics. 2017 Mar 20;18(1):178. doi: 10.1186/s12859-017-1592-1.
Using phylogenomic analysis tools for tracking pathogens has become standard practice in academia, public health agencies, and large industries. Using the same raw read genomic data as input, there are several different approaches being used to infer phylogenetic tree. These include many different SNP pipelines, wgMLST approaches, k-mer algorithms, whole genome alignment and others; each of these has advantages and disadvantages, some have been extensively validated, some are faster, some have higher resolution. A few of these analysis approaches are well-integrated into the regulatory process of US Federal agencies (e.g. the FDA's SNP pipeline for tracking foodborne pathogens). However, despite extensive validation on benchmark datasets and comparison with other pipelines, we lack methods for fully exploring the effects of multiple parameter values in each pipeline that can potentially have an effect on whether the correct phylogenetic tree is recovered.
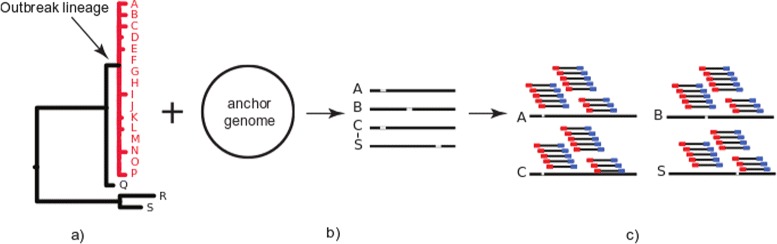
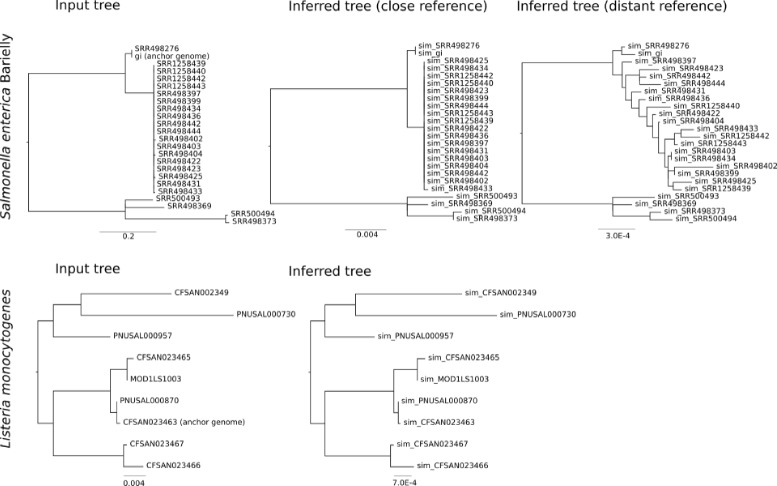
To resolve this problem, we offer a program, TreeToReads, which can generate raw read data from mutated genomes simulated under a known phylogeny. This simulation pipeline allows direct comparisons of simulated and observed data in a controlled environment. At each step of these simulations, researchers can vary parameters of interest (e.g., input tree topology, amount of sequence divergence, rate of indels, read coverage, distance of reference genome, etc) to assess the effects of various parameter values on correctly calling SNPs and reconstructing an accurate tree.
Such critical assessments of the accuracy and robustness of analytical pipelines are essential to progress in both research and applied settings.
使用系统发育基因组分析工具追踪病原体已成为学术界、公共卫生机构和大型企业的标准做法。使用相同的原始读取基因组数据作为输入,有几种不同的方法用于推断系统发育树。这些方法包括许多不同的单核苷酸多态性(SNP)管道、全基因组多位点序列分型(wgMLST)方法、k-mer算法、全基因组比对等;每种方法都有优缺点,有些已得到广泛验证,有些速度更快,有些分辨率更高。其中一些分析方法已很好地融入美国联邦机构的监管流程(例如,美国食品药品监督管理局(FDA)用于追踪食源性病原体的SNP管道)。然而,尽管在基准数据集上进行了广泛验证并与其他管道进行了比较,但我们缺乏全面探索每个管道中多个参数值的影响的方法,这些参数值可能会影响是否能恢复正确的系统发育树。
为了解决这个问题,我们提供了一个程序TreeToReads,它可以从在已知系统发育下模拟的突变基因组中生成原始读取数据。这个模拟管道允许在可控环境中直接比较模拟数据和观测数据。在这些模拟的每个步骤中,研究人员可以改变感兴趣的参数(例如,输入树拓扑结构、序列分歧量、插入缺失率、读取覆盖率、参考基因组距离等),以评估各种参数值对正确调用SNP和重建准确树的影响。
对分析管道的准确性和稳健性进行此类关键评估对于研究和应用环境的进展至关重要。