1School of Life Science and Technology, Tokyo Institute of Technology, Tokyo, Japan.
2Department of Bacteriology, Faculty of Medical Sciences, Kyushu University, Fukuoka, Japan.
Microb Genom. 2019 May;5(5). doi: 10.1099/mgen.0.000261. Epub 2019 May 17.
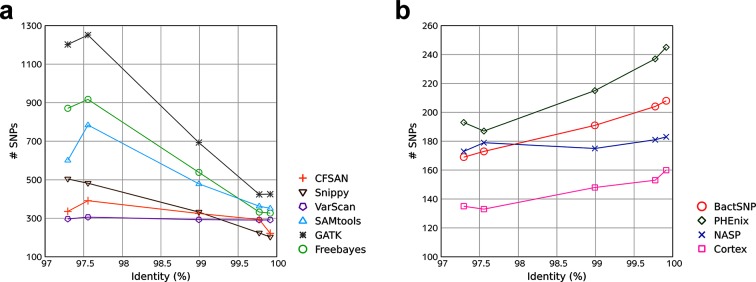
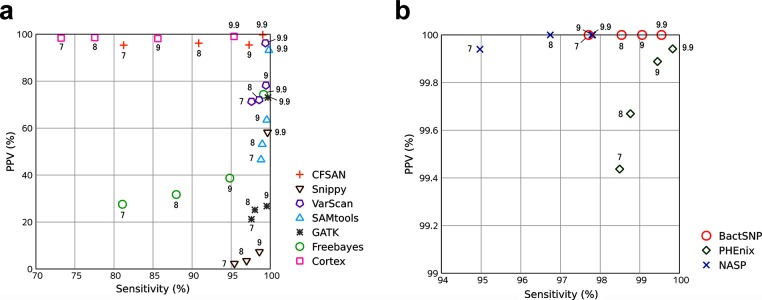
Bacteria are highly diverse, even within a species; thus, there have been many studies which classify a single species into multiple types and analyze the genetic differences between them. Recently, the use of whole-genome sequencing (WGS) has been popular for these analyses, and the identification of single-nucleotide polymorphisms (SNPs) between isolates is the most basic analysis performed following WGS. The performance of SNP-calling methods therefore has a significant effect on the accuracy of downstream analyses, such as phylogenetic tree inference. In particular, when closely related isolates are analyzed, e.g. in outbreak investigations, some SNP callers tend to detect a high number of false-positive SNPs compared with the limited number of true SNPs among isolates. However, the performances of various SNP callers in such a situation have not been validated sufficiently. Here, we show the results of realistic benchmarks of commonly used SNP callers, revealing that some of them exhibit markedly low accuracy when target isolates are closely related. As an alternative, we developed a novel pipeline BactSNP, which utilizes both assembly and mapping information and is capable of highly accurate and sensitive SNP calling in a single step. BactSNP is also able to call SNPs among isolates when the reference genome is a draft one or even when the user does not input the reference genome. BactSNP is available at https://github.com/IEkAdN/BactSNP.
细菌具有高度的多样性,即使在同一物种内也是如此;因此,已经有许多研究将单一物种分为多种类型,并分析它们之间的遗传差异。最近,全基因组测序(WGS)已广泛用于这些分析,并且在 WGS 之后,对分离株之间的单核苷酸多态性(SNP)进行鉴定是最基本的分析。因此,SNP 调用方法的性能对下游分析(如系统发育树推断)的准确性有重大影响。特别是在分析密切相关的分离株时,例如在暴发调查中,与分离株之间有限数量的真正 SNP 相比,一些 SNP 调用者倾向于检测到大量假阳性 SNP。然而,在这种情况下,各种 SNP 调用者的性能尚未得到充分验证。在这里,我们展示了常用 SNP 调用者的现实基准测试结果,表明当目标分离株密切相关时,其中一些调用者的准确性明显较低。作为替代方案,我们开发了一种新的 SNP 调用流水线 BactSNP,它利用组装和映射信息,能够在单个步骤中实现高度准确和敏感的 SNP 调用。BactSNP 还能够在参考基因组是草图甚至用户未输入参考基因组的情况下,在分离株之间调用 SNP。BactSNP 可在 https://github.com/IEkAdN/BactSNP 上获得。