Brown Bonnie L, Watson Mick, Minot Samuel S, Rivera Maria C, Franklin Rima B
Virginia Commonwealth University, Department of Biology, 1000 W Cary Street, Richmond, VA 23284, USA.
The Roslin Institute, University of Edinburgh, Division of Genetics and Genomics, Easter Bush, Midlothian, EH25 9RG, UK.
Gigascience. 2017 Mar 1;6(3):1-10. doi: 10.1093/gigascience/gix007.
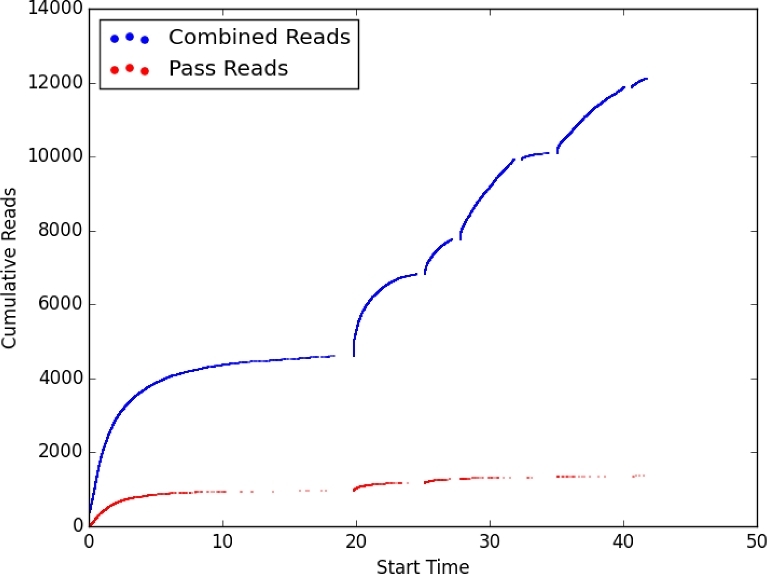
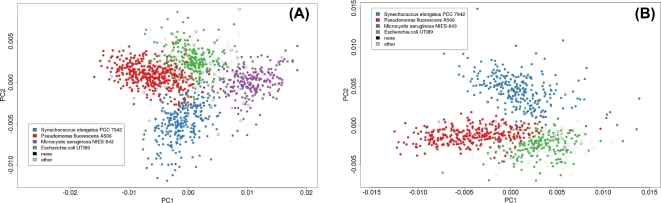
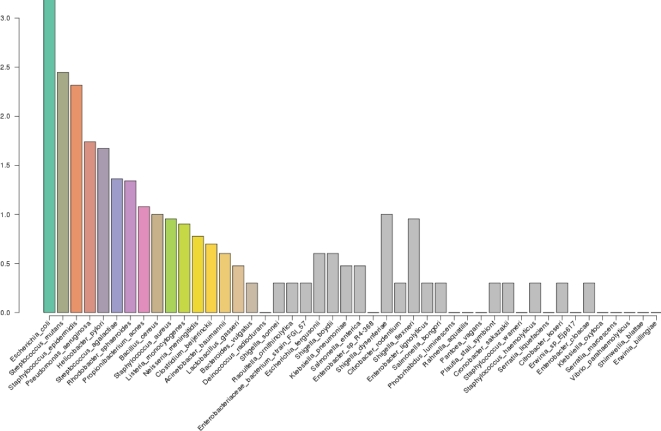
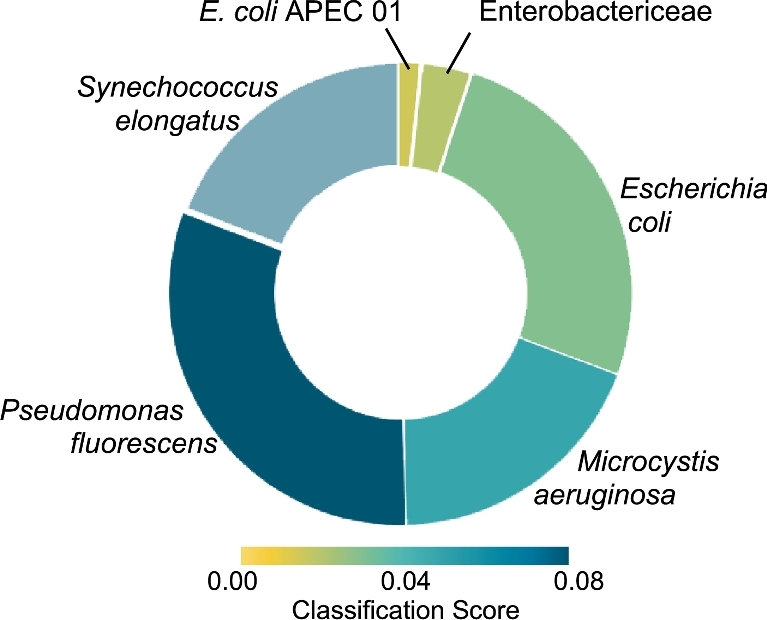
Environmental metagenomic analysis is typically accomplished by assigning taxonomy and/or function from whole genome sequencing or 16S amplicon sequences. Both of these approaches are limited, however, by read length, among other technical and biological factors. A nanopore-based sequencing platform, MinION™, produces reads that are ≥1 × 104 bp in length, potentially providing for more precise assignment, thereby alleviating some of the limitations inherent in determining metagenome composition from short reads. We tested the ability of sequence data produced by MinION (R7.3 flow cells) to correctly assign taxonomy in single bacterial species runs and in three types of low-complexity synthetic communities: a mixture of DNA using equal mass from four species, a community with one relatively rare (1%) and three abundant (33% each) components, and a mixture of genomic DNA from 20 bacterial strains of staggered representation. Taxonomic composition of the low-complexity communities was assessed by analyzing the MinION sequence data with three different bioinformatic approaches: Kraken, MG-RAST, and One Codex. Results: Long read sequences generated from libraries prepared from single strains using the version 5 kit and chemistry, run on the original MinION device, yielded as few as 224 to as many as 3497 bidirectional high-quality (2D) reads with an average overall study length of 6000 bp. For the single-strain analyses, assignment of reads to the correct genus by different methods ranged from 53.1% to 99.5%, assignment to the correct species ranged from 23.9% to 99.5%, and the majority of misassigned reads were to closely related organisms. A synthetic metagenome sequenced with the same setup yielded 714 high quality 2D reads of approximately 5500 bp that were up to 98% correctly assigned to the species level. Synthetic metagenome MinION libraries generated using version 6 kit and chemistry yielded from 899 to 3497 2D reads with lengths averaging 5700 bp with up to 98% assignment accuracy at the species level. The observed community proportions for “equal” and “rare” synthetic libraries were close to the known proportions, deviating from 0.1% to 10% across all tests. For a 20-species mock community with staggered contributions, a sequencing run detected all but 3 species (each included at <0.05% of DNA in the total mixture), 91% of reads were assigned to the correct species, 93% of reads were assigned to the correct genus, and >99% of reads were assigned to the correct family. Conclusions: At the current level of output and sequence quality (just under 4 × 103 2D reads for a synthetic metagenome), MinION sequencing followed by Kraken or One Codex analysis has the potential to provide rapid and accurate metagenomic analysis where the consortium is comprised of a limited number of taxa. Important considerations noted in this study included: high sensitivity of the MinION platform to the quality of input DNA, high variability of sequencing results across libraries and flow cells, and relatively small numbers of 2D reads per analysis limit. Together, these limited detection of very rare components of the microbial consortia, and would likely limit the utility of MinION for the sequencing of high-complexity metagenomic communities where thousands of taxa are expected. Furthermore, the limitations of the currently available data analysis tools suggest there is considerable room for improvement in the analytical approaches for the characterization of microbial communities using long reads. Nevertheless, the fact that the accurate taxonomic assignment of high-quality reads generated by MinION is approaching 99.5% and, in most cases, the inferred community structure mirrors the known proportions of a synthetic mixture warrants further exploration of practical application to environmental metagenomics as the platform continues to develop and improve. With further improvement in sequence throughput and error rate reduction, this platform shows great promise for precise real-time analysis of the composition and structure of more complex microbial communities.
环境宏基因组分析通常通过对全基因组测序或16S扩增子序列进行分类和/或功能分配来完成。然而,这两种方法都受到读取长度以及其他技术和生物学因素的限制。基于纳米孔的测序平台MinION™产生长度≥1×104 bp的读取序列,有可能提供更精确的分配,从而减轻从短读取序列确定宏基因组组成时固有的一些限制。我们测试了MinION(R7.3流动槽)产生的序列数据在单一细菌物种运行以及三种低复杂性合成群落中正确进行分类的能力:一种由来自四个物种的等量DNA组成的混合物、一个具有一种相对罕见(1%)和三种丰富(各33%)成分的群落,以及一个来自20个细菌菌株的交错代表性基因组DNA混合物。通过使用三种不同的生物信息学方法(Kraken、MG-RAST和One Codex)分析MinION序列数据,评估低复杂性群落的分类组成。结果:使用第5版试剂盒和化学方法从单菌株制备的文库中生成的长读取序列,在原始MinION设备上运行,产生的双向高质量(2D)读取序列少至224条至多至3497条,整个研究的平均长度为6000 bp。对于单菌株分析,不同方法将读取序列正确分配到正确属的比例范围为53.1%至99.5%,分配到正确物种的比例范围为23.9%至99.5%,大多数错误分配的读取序列是分配给了密切相关的生物体。以相同设置测序的合成宏基因组产生了714条高质量的约5500 bp的2D读取序列,其中高达98%在物种水平上被正确分配。使用第6版试剂盒和化学方法生成的合成宏基因组MinION文库产生了899至3497条2D读取序列,长度平均为5700 bp,在物种水平上的分配准确率高达98%。在所有测试中,“等量”和“罕见”合成文库中观察到的群落比例接近已知比例,偏差为0.1%至10%。对于一个具有交错贡献的20物种模拟群落,一次测序运行检测到了除3个物种(每个物种在总混合物中的DNA含量<0.05%)之外的所有物种,91%的读取序列被分配到正确的物种,93%的读取序列被分配到正确的属,>99%的读取序列被分配到正确的科。结论:在当前的输出水平和序列质量(合成宏基因组约4×103条2D读取序列)下,MinION测序随后进行Kraken或One Codex分析有潜力在群落由有限数量的分类群组成时提供快速准确的宏基因组分析。本研究中指出的重要考虑因素包括:MinION平台对输入DNA质量的高敏感性、不同文库和流动槽之间测序结果的高变异性,以及每次分析中相对较少的2D读取序列数量限制。这些因素共同限制了对微生物群落中非常罕见成分的检测,并可能限制MinION在预期有数千个分类群的高复杂性宏基因组群落测序中的效用。此外,当前可用数据分析工具的局限性表明,使用长读取序列表征微生物群落的分析方法有很大的改进空间。然而,MinION产生的高质量读取序列的准确分类分配接近99.5%,并且在大多数情况下,推断的群落结构反映了合成混合物的已知比例,这一事实值得随着该平台不断发展和改进,进一步探索其在环境宏基因组学中的实际应用。随着序列通量的进一步提高和错误率的降低,该平台对于更复杂微生物群落的组成和结构进行精确实时分析显示出巨大的前景。