Lustgarten Jonathan Lyle, Balasubramanian Jeya Balaji, Visweswaran Shyam, Gopalakrishnan Vanathi
Red Bank Veterinary Hospital / 2051 Briggs Rd, Mt Laurel, NJ 08054, USA.
Intelligent Systems Program, University of Pittsburgh / 5113 Sennott Square, 210 South Bouquet Street, Pittsburgh, PA 15260, USA.
Data (Basel). 2017 Mar;2(1). doi: 10.3390/data2010005. Epub 2017 Jan 18.
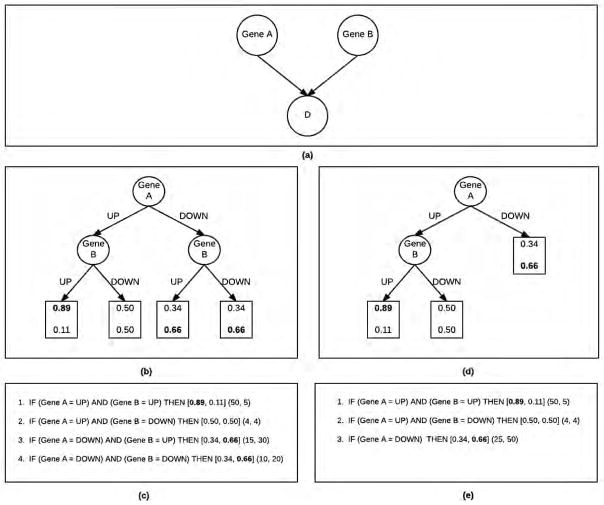
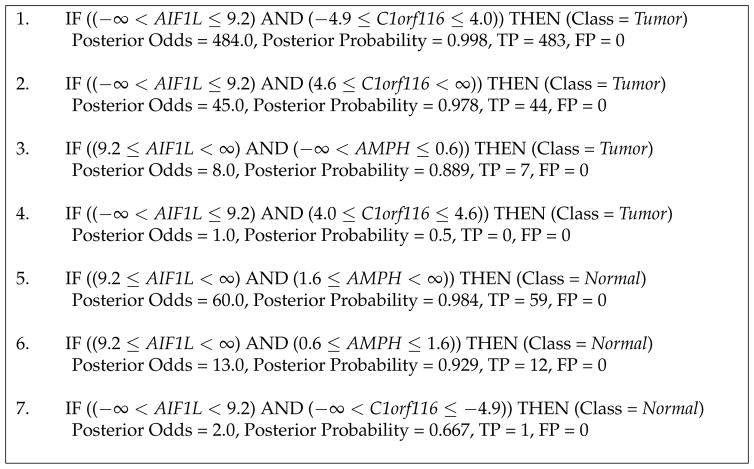
The comprehensibility of good predictive models learned from high-dimensional gene expression data is attractive because it can lead to biomarker discovery. Several good classifiers provide comparable predictive performance but differ in their abilities to summarize the observed data. We extend a Bayesian Rule Learning (BRL-GSS) algorithm, previously shown to be a significantly better predictor than other classical approaches in this domain. It searches a space of Bayesian networks using a decision tree representation of its parameters with global constraints, and infers a set of IF-THEN rules. The number of parameters and therefore the number of rules are combinatorial to the number of predictor variables in the model. We relax these global constraints to a more generalizable local structure (BRL-LSS). BRL-LSS entails more parsimonious set of rules because it does not have to generate all combinatorial rules. The search space of local structures is much richer than the space of global structures. We design the BRL-LSS with the same worst-case time-complexity as BRL-GSS while exploring a richer and more complex model space. We measure predictive performance using Area Under the ROC curve (AUC) and Accuracy. We measure model parsimony performance by noting the average number of rules and variables needed to describe the observed data. We evaluate the predictive and parsimony performance of BRL-GSS, BRL-LSS and the state-of-the-art C4.5 decision tree algorithm, across 10-fold cross-validation using ten microarray gene-expression diagnostic datasets. In these experiments, we observe that BRL-LSS is similar to BRL-GSS in terms of predictive performance, while generating a much more parsimonious set of rules to explain the same observed data. BRL-LSS also needs fewer variables than C4.5 to explain the data with similar predictive performance. We also conduct a feasibility study to demonstrate the general applicability of our BRL methods on the newer RNA sequencing gene-expression data.
从高维基因表达数据中学习到的良好预测模型的可理解性很有吸引力,因为它可以促成生物标志物的发现。几个性能良好的分类器提供了相当的预测性能,但在总结观测数据的能力方面有所不同。我们扩展了一种贝叶斯规则学习(BRL-GSS)算法,该算法此前已被证明在该领域是比其他经典方法显著更好的预测器。它使用带有全局约束的参数决策树表示来搜索贝叶斯网络空间,并推断出一组“如果……那么……”规则。参数的数量以及因此规则的数量与模型中预测变量的数量是组合关系。我们将这些全局约束放宽到更具通用性的局部结构(BRL-LSS)。BRL-LSS需要的规则集更为简洁,因为它不必生成所有组合规则。局部结构的搜索空间比全局结构的空间丰富得多。我们设计的BRL-LSS与BRL-GSS具有相同的最坏情况时间复杂度,同时探索更丰富、更复杂的模型空间。我们使用ROC曲线下面积(AUC)和准确率来衡量预测性能。我们通过记录描述观测数据所需的规则和变量的平均数量来衡量模型的简洁性性能。我们使用十个微阵列基因表达诊断数据集,通过10折交叉验证来评估BRL-GSS、BRL-LSS和最先进的C4.5决策树算法的预测性能和简洁性性能。在这些实验中,我们观察到BRL-LSS在预测性能方面与BRL-GSS相似,同时生成一组简洁得多的规则来解释相同的观测数据。BRL-LSS在具有相似预测性能的情况下,解释数据所需的变量也比C4.5少。我们还进行了一项可行性研究,以证明我们的BRL方法在更新的RNA测序基因表达数据上的普遍适用性。