Musicha Patrick, Feasey Nicholas A, Cain Amy K, Kallonen Teemu, Chaguza Chrispin, Peno Chikondi, Khonga Margaret, Thompson Sarah, Gray Katherine J, Mather Alison E, Heyderman Robert S, Everett Dean B, Thomson Nicholas R, Msefula Chisomo L
Malawi-Liverpool-Wellcome Trust Clinical Research Programme, Queen Elizabeth Central Hospital, Blantyre, Malawi.
Liverpool School of Tropical Medicine, Liverpool, UK.
J Antimicrob Chemother. 2017 Jun 1;72(6):1602-1609. doi: 10.1093/jac/dkx058.
Efforts to treat Escherichia coli infections are increasingly being compromised by the rapid, global spread of antimicrobial resistance (AMR). Whilst AMR in E. coli has been extensively investigated in resource-rich settings, in sub-Saharan Africa molecular patterns of AMR are not well described. In this study, we have begun to explore the population structure and molecular determinants of AMR amongst E. coli isolates from Malawi.
Ninety-four E. coli isolates from patients admitted to Queen's Hospital, Malawi, were whole-genome sequenced. The isolates were selected on the basis of diversity of phenotypic resistance profiles and clinical source of isolation (blood, CSF and rectal swab). Sequence data were analysed using comparative genomics and phylogenetics.
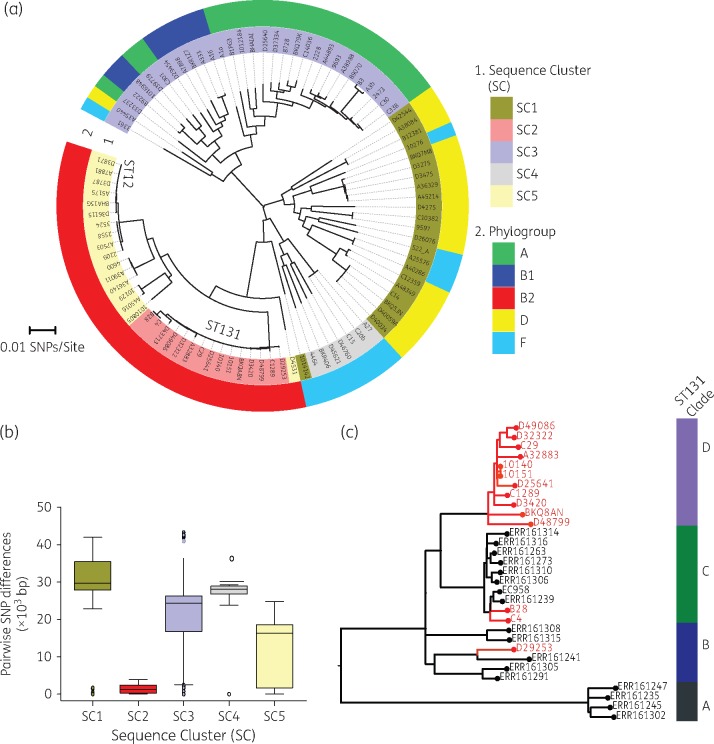
Our results revealed the presence of five clades, which were strongly associated with E. coli phylogroups A, B1, B2, D and F. We identified 43 multilocus STs, of which ST131 (14.9%) and ST12 (9.6%) were the most common. We identified 25 AMR genes. The most common ESBL gene was bla CTX-M-15 and it was present in all five phylogroups and 11 STs, and most commonly detected in ST391 (4/4 isolates), ST648 (3/3 isolates) and ST131 [3/14 (21.4%) isolates].
This study has revealed a high diversity of lineages associated with AMR, including ESBL and fluoroquinolone resistance, in Malawi. The data highlight the value of longitudinal bacteraemia surveillance coupled with detailed molecular epidemiology in all settings, including low-income settings, in describing the global epidemiology of ESBL resistance.
抗菌药物耐药性(AMR)在全球迅速传播,正日益影响大肠杆菌感染的治疗效果。虽然在资源丰富的地区对大肠杆菌中的AMR进行了广泛研究,但在撒哈拉以南非洲,AMR的分子模式尚未得到充分描述。在本研究中,我们开始探索马拉维大肠杆菌分离株中AMR的种群结构和分子决定因素。
对马拉维皇后医院收治患者的94株大肠杆菌分离株进行全基因组测序。根据表型耐药谱的多样性和分离的临床来源(血液、脑脊液和直肠拭子)选择分离株。使用比较基因组学和系统发育学分析序列数据。
我们的结果显示存在五个进化枝,它们与大肠杆菌菌系A、B1、B2、D和F密切相关。我们鉴定出43个多位点序列类型(MLST),其中ST131(14.9%)和ST12(9.6%)最为常见。我们鉴定出25个AMR基因。最常见的超广谱β-内酰胺酶(ESBL)基因是bla CTX-M-15,它存在于所有五个菌系和11个ST中,最常见于ST391(4/4株分离株)、ST648(3/3株分离株)和ST131 [3/14(21.4%)株分离株]。
本研究揭示了马拉维与AMR相关的谱系具有高度多样性,包括ESBL和氟喹诺酮耐药性。这些数据突出了在包括低收入环境在内的所有环境中,进行纵向菌血症监测并结合详细分子流行病学对于描述ESBL耐药性全球流行病学的价值。